2021 - Present
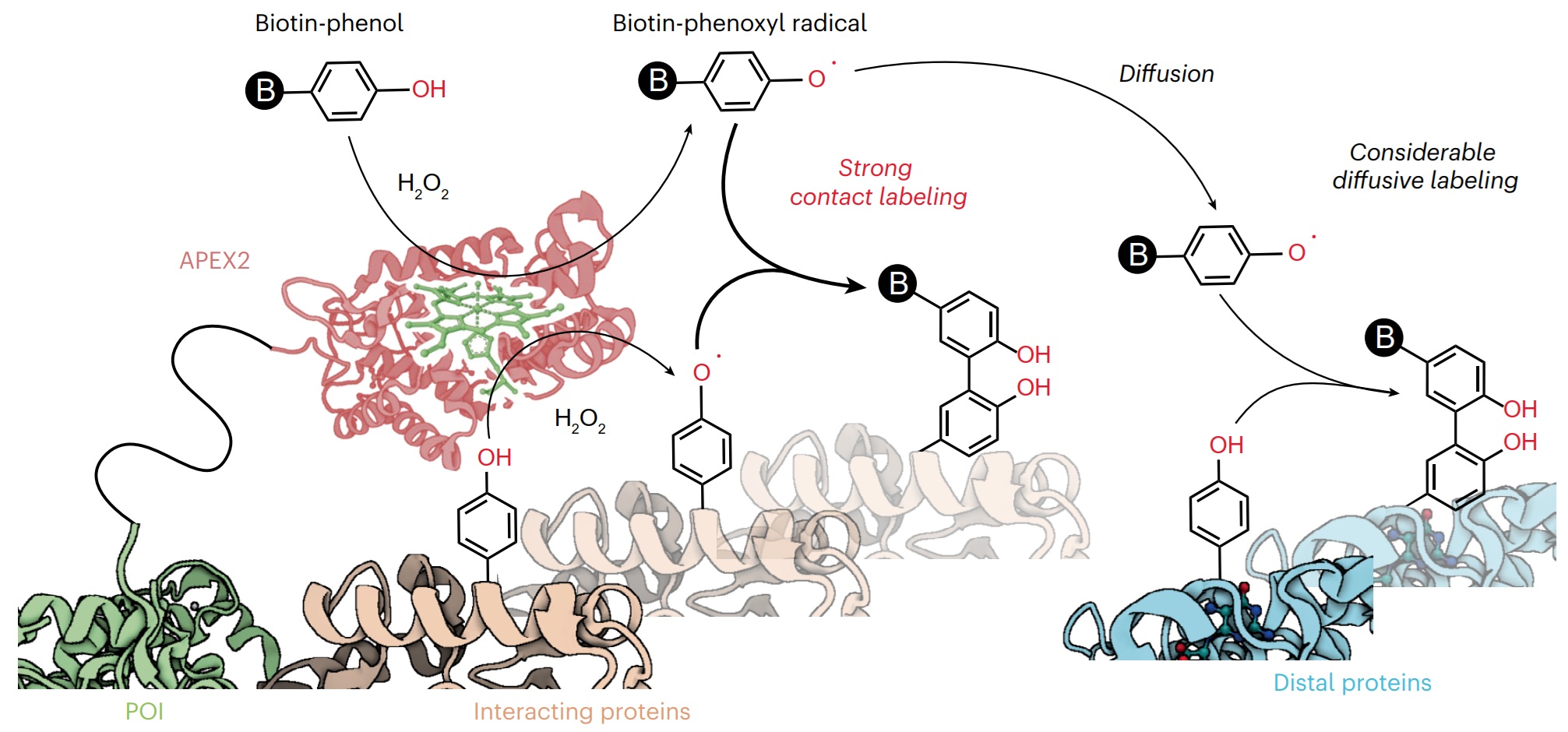
Proximity labeling techniques such as TurboID and APEX2 have become pivotal tools for studying protein interactions. However, the spatial patterns of labeling methods within the submicrometer range remain poorly understood. Here we used DNA nanostructure platforms to precisely measure the labeling radii of TurboID and APEX2 through in vitro assays. Our DNA nanoruler design enables the deployment of oligonucleotide-barcoded labeling targets with nanometer precision near the enzymes. By quantifying labeling yields using qPCR and mapping them against target distances, we uncovered surprising insights into the labeling mechanisms. Contrary to the prevailing diffusive labeling model, our results demonstrate that TurboID primarily operates through contact-dependent labeling. Similarly, APEX2 shows high labeling efficiency within its direct contact range. In parallel, it exhibits low-level diffusive labeling toward more distant phenols. These findings reframe our understanding in the mechanism of proximity labeling enzymes while highlighting the potential of DNA nanotechnology in spatially profiling reactive species.
Yang, Z., Zhang, Y., Fang, Y., Zhang, Y., Du, J., Shen, X., Zhang, K., Zou, P.* and Chen, Z.* (2025). Nat. Chem. Biol. In press.
In this issue of Neuron, Grimm et al. describe the Jarvis voltage indicator, which demonstrates that scanless parallel two-photon excitation enables robust, high-speed voltage imaging in vivo, overturning prior assumptions about the incompatibility of rhodopsin-based indicators with two-photon microscopy.
Liu, S.#, Peng, L.# and Zou, P.* (2026). Neuron 114, 1163-1165.
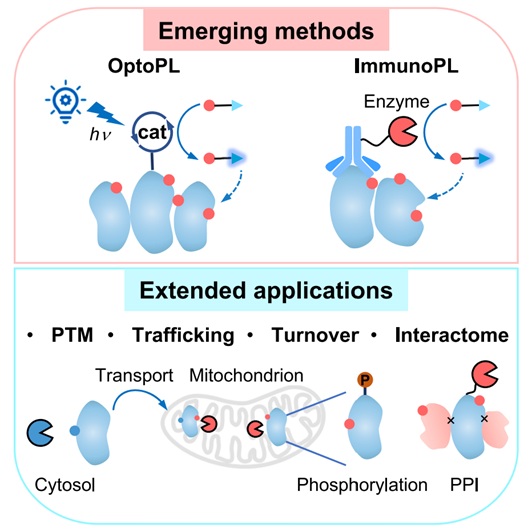
The spatial organization of the cellular proteome is vital for cellular physiology, as protein localization is closely linked to post-translational modifications, subcellular trafficking, and protein-protein interactions. Systematic profiling of these spatial features can greatly enhance our understanding of protein functions. Recent advances in enzyme-mediated proximity labeling (PL) techniques, such as TurboID and APEX2, have improved our ability to map subcellular proteomes in living cells. This review discusses emerging trends in PL methods, which now offer subcellular precision with multi-dimensional protein features, including post-translational modifications, trafficking, turnover, and interaction with other biomolecules. Additionally, new techniques such as photoactivatable PL (optoPL) and antibody-targeted PL (immunoPL) provide enhanced spatiotemporal control and allow for detailed subcellular proteome mapping without genetic manipulation.
Wang, G.#, Liu, J.#, Sun, X.#, Qin, W.*, Han, S.* and Zou, P.* (2026). Mol. Cell Proteomics 25, 101520.
The increasing prevalence of emerging infectious diseases highlights the urgent need for broad-spectrum antiviral therapeutics. Here, the discovery of SB2960, a small molecule, is reported that promotes stress granule (SG) formation and exhibits potent antiviral activity across diverse viral species. SB2960 suppresses viral replication with minimal cytotoxicity by modulating host antiviral immune responses. Target identification studies reveal that SB2960 engages receptor for activated protein C kinase 1 (RACK1), a key regulator of SG dynamics. Integrated transcriptomic and proteomic analyses further demonstrate that SB2960 alters the composition of SG and modulates the expression of antiviral genes. These findings establish SB2960 as a promising host-directed broad-spectrum antiviral agent and a valuable probe for investigating SG biology in the context of viral infection.
Byun, W. G.#, Son, S.#, Lee, J., Lee, M., Ko, M., Zhao, S., Kim, J. H., Vaithegi, K., Kwon, J. H., Lee, J. H., Jeon, S., Yi, D., Zou, P., Kim, S.* and Park, S. B.* (2025). Adv. Sci. 13, e12972.
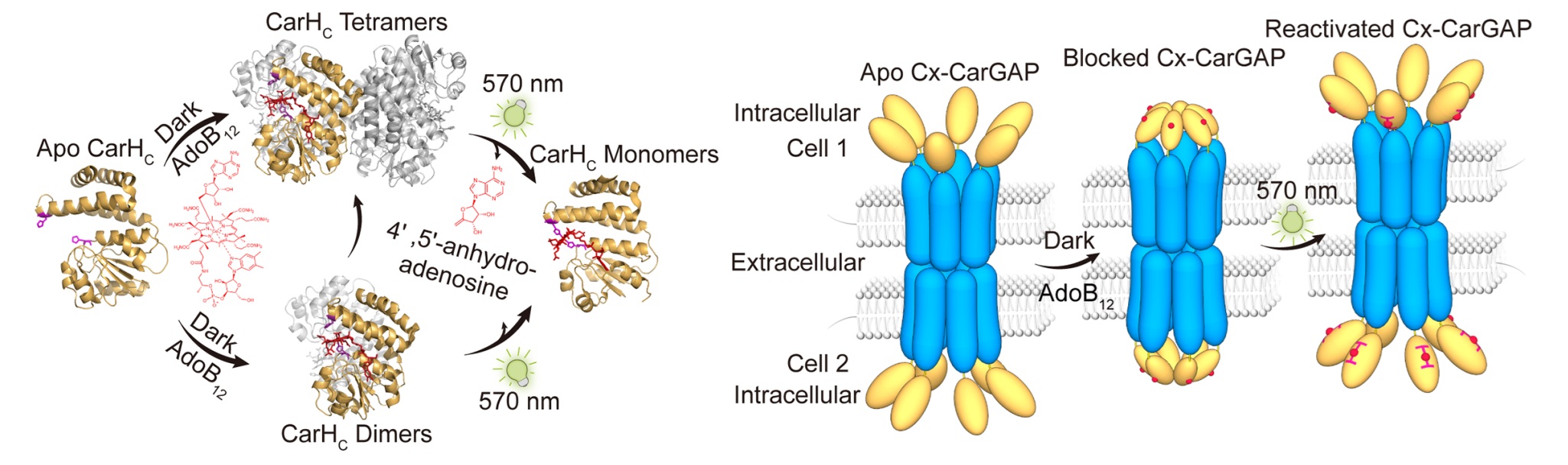
Gap junctions mediate rapid signal transduction between contiguous cells, which are indispensable for multicellular organisms to coordinate cellular activities across numerous physiological processes. However, precise control of gap junctions remains elusive. Herein, we present CarGAP, a single-component chemo-optogenetic tool that utilizes the C-terminal adenosylcobalamin (AdoB12) binding domain of a photoreceptor protein (i.e., CarHC) to achieve reversible control over both vertebrate and invertebrate gap junctions with spatiotemporal precision. The vertebrate CarGAP (i.e., Cx-CarGAP), created by genetically fusing connexins with CarHC in mammalian cells, can efficiently block the gap junction channels through AdoB12-induced protein oligomerization and subsequently reinstate them via green light-induced protein disassembly. We further introduced the CarGAP system (i.e., Inx-CarGAP) to the Drosophila ovary, enabling reversible control over the heterotypic gap junctions formed by innexin2 (Inx2) and innexin4 (Inx4, also known as zero population growth, Zpg), thereby uncovering the roles of gap junctions in stem cell-niche interactions. This study illustrates CarGAP as a generalizable chemo-optogenetic tool for interrogating the functions of gap junctions in various biological contexts.
Cui, D.#, Liu, S.#, Huang, X.#, Tang, X. A., Zheng, M., He, Z., Xu, R., Sun, C., Xu, Y., Tu, R.*, Zou, P.*, Xie, T.* and Sun, F.* (2025). Proc. Natl. Acad. Sci. U. S. A. 122, e2518037122.
Cell-cell communication (CCC) is fundamental to essential biological processes including growth, differentiation, immune surveillance, and tissue homeostasis, and its dysregulation underlies various diseases such as cancer, autoimmunity, and neurodegeneration. In response to growing interest in decoding complex multicellular interactions, the 380th Shuangqing Forum entitled "Chemical, Biological, and Medical Frontiers in Multicellular Complex Systems" was convened, providing a platform to discuss recent interdisciplinary breakthroughs. This review, emerging from forum discussions, highlights the latest advancements in molecular tools—such as super-resolution imaging, proximity labeling, bioorthogonal chemistry, synthetic receptors, and single-cell spatial omics—that enable unprecedented insights into spatial, molecular, and functional aspects of CCC. Emphasizing their translational potential, we discuss their profound implications for immuno-oncology, regenerative medicine, and autoimmune diseases. We further outline current challenges and opportunities, particularly advocating for a future precision medicine framework centered around targeted modulation of cell-cell interactions.
Li, J. P., Guo, W., Zou, P., Chu, C., Wu, J., Guo, Z., Huang, Y.*, Yang, J.*, Chen, P. R.* (2025). Natl. Sci. Rev. 12, nwaf473.
Proteomics is transforming medical sciences, but bridging isolated samples with intact in vivo microenvironments remains a major hurdle. We present an in vivo proteomic labeling (IVPL) platform built on a new substrate, Btn-Ph-3F, and engineered ascorbate peroxidase (APEX2)-EGFPf/f mice. Btn-Ph-3F shows high stability in organs possessing complex microenvironments, while APEX2-EGFPf/f mice readily cross with commercial Cre lines, enabling specific proteomic labeling for customized cell groups in distant organs. IVPL robustly profiles in situ proteomes of intestinal epithelium, mammary gland, and tumor-infiltrating Treg cells, and, critically, labels trace exogenous proteomes from patient-derived exosomes in live mice. We identify lactate dehydrogenase A-like 6A (LDHAL6A) as a persisting exosomal effector that promotes malignant programs in recipient cells. Inhibition of LDHAL6A combined with paclitaxel treatment markedly suppresses triple-negative breast cancer growth and metastasis. Collectively, our work not only establishes an advanced model for IVPL but also profiles ultimately exosomal actors in recipient organs for targeted therapy.
Wang, Q.#, Jiang, Y.#, Bi, M.#, Wang, G., Xiao, Y., Zhao, H., Guo, Y., Li, X., Yue, W., Zhang, N., Xie, B., Xue, Y., Yin, H., Zou, P. and Li, M.* (2025). Mol. Cell 85, 4651-4666.
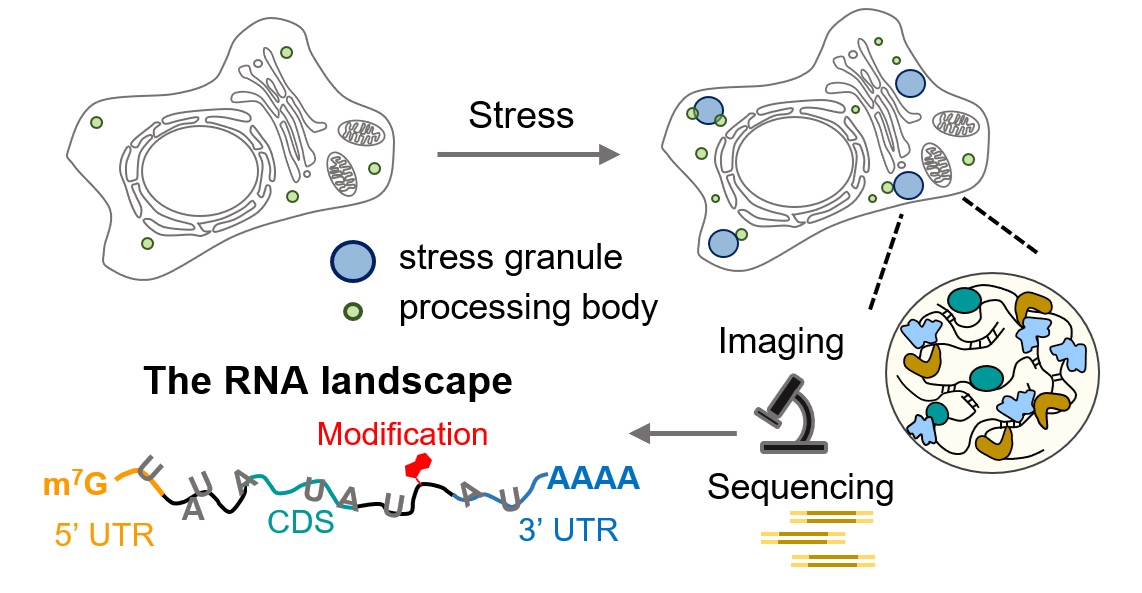
RNA granules, including stress granules (SGs) and processing bodies (PBs), are membraneless organelles that orchestrate RNA localization, metabolism, and translational control in response to cellular stress. This Perspective examines the RNA landscape within SGs and PBs, highlighting recent insights into how these compartments shape RNA fate. We review current methodologies for probing granule-associated RNAs, including high-resolution imaging, transcriptomics, and sequencing-based approaches. In particular, we spotlight emerging photoactivated proximity labeling techniques that offer unprecedented spatiotemporal resolution for mapping RNA interactions in living cells. Looking ahead, we propose combining multiomic approaches to better define the roles of RNAs within granules. Finally, we emphasize the importance of investigating RNA granules in neuronal contexts, where dysregulated RNA condensation is intimately linked to neurodegenerative diseases. Together, these approaches promise to elucidate the molecular logic by which RNA granules govern post-transcriptional gene regulation and cellular adaptation to stress.
Ren, Z.#, Zhao, S.# and Zou, P.* (2025). Biochemistry 64, 3156-3164.
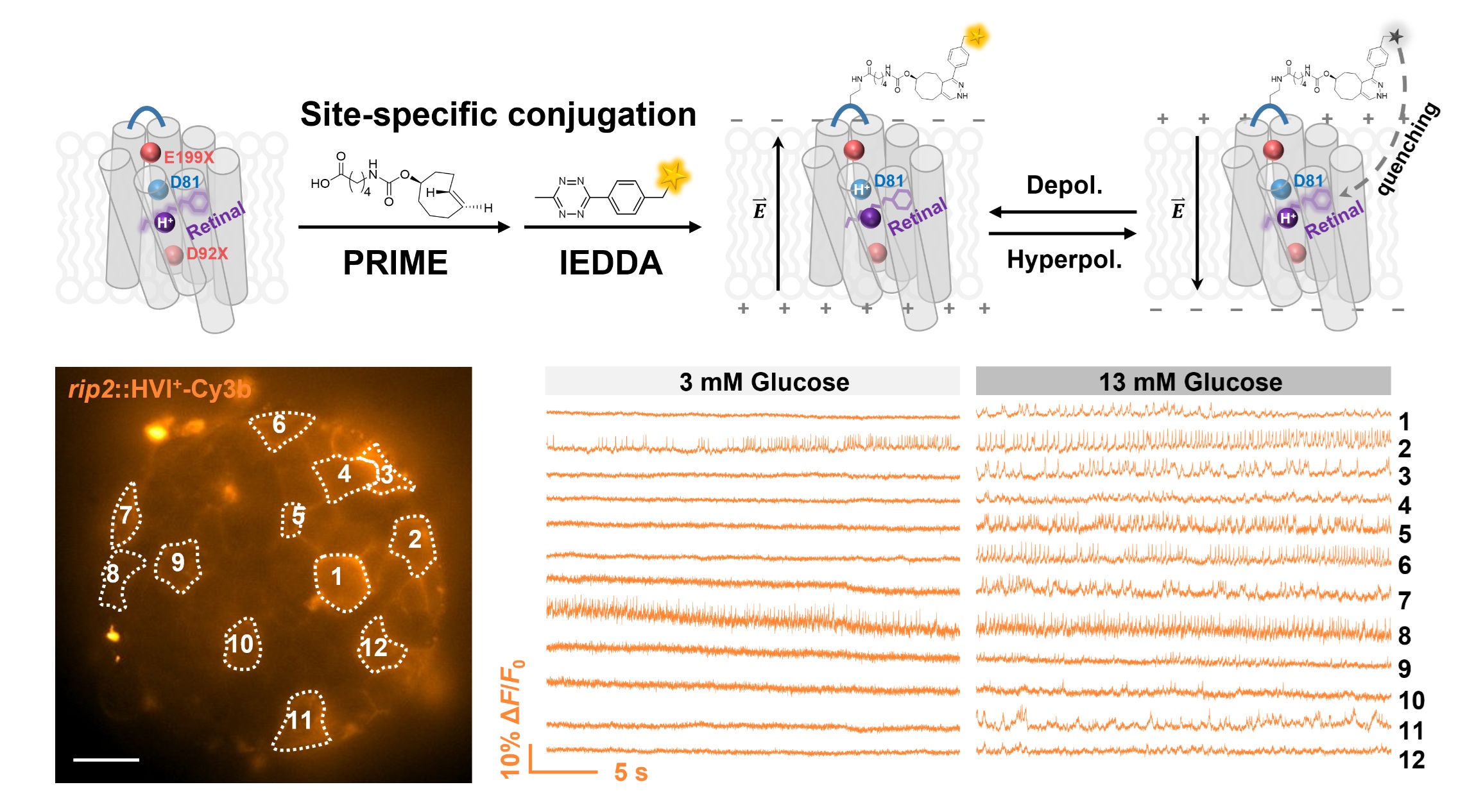
Membrane potential dynamics are crucial to the physiology of electrically excitable cells, such as neurons, cardiomyocytes, and pancreatic islet cells. Fluorescence voltage indicators have enabled the visualization of electrical activities at the single-cell level across large cell populations. However, most existing indicators have a negative fluorescence-voltage response, resulting in a high background signal when cells are at their resting state. To reduce background interference during voltage imaging, we developed a positive-going hybrid voltage indicator (HVI+), which has lower intensity at the resting state and becomes brighter upon membrane depolarization. HVI+-Cy3b exhibits a remarkable voltage sensitivity of 55% ΔF/F0 per 100 mV, enabling accurate reporting of action potential waveforms in beating cardiomyocytes and neurons. In addition, by combining HVI+-Cy3b with HVI-Cy3b, we demonstrate the capability to simultaneously assess the effects of glucose on the spike activities of two cell types in pancreatic islets.
Liu, S.#, Ling, J.#, Xie, B.#, Zhang, Y., Peng, L., Yang, L., Yu, L., Lin, J., Tang, C.*, Chen, Z.* and Zou, P.* (2025). Sci. Adv. 11, eads1807.
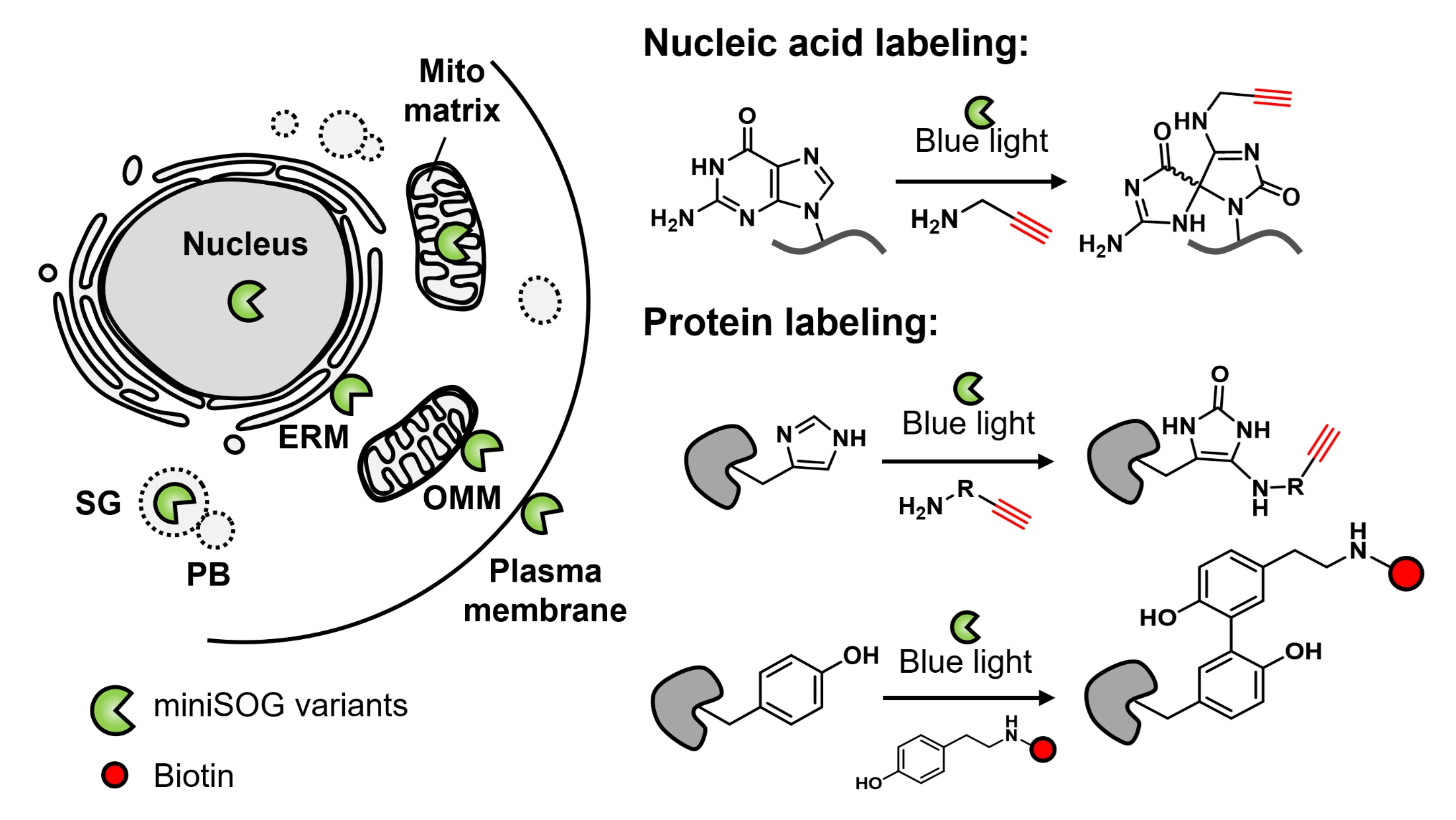
Engineered photosensitizer proteins, such as miniSOG, KillerRed, and SuperNova, have long been used for light-triggered protein inhibition and cell ablation. Compared to synthetic organic dyes, these genetically encoded tags provide superior spatial precision for subcellular targeting. More recently, the photochemistry of miniSOG has been repurposed for subcellular omics studies. Upon light activation, miniSOG generates reactive oxygen species (ROS) that oxidize nearby nucleic acids or proteins. These oxidized biomolecules can then react with exogenously supplied nucleophilic probes, which introduce bio-orthogonal handles for downstream enrichment and analysis. This labeling strategy, known as photocatalytic proximity labeling (PPL), has emerged as a powerful approach for profiling the molecular architecture of subcellular compartments and identifying RNA or protein interactors of specific targets. The use of light provides exceptional temporal control, enabling labeling windows as short as 1 s. Moreover, PPL readily supports pulse-chase experiments through simple light on/off switching, an advantage not easily achievable with conventional platforms such as APEX or TurboID. In this account, we highlight our recent developments and applications of genetically encoded PPL tools. These include CAP-seq for RNA/DNA labeling, RinID for protein labeling, and LAP-seq/MS/CELL for bioluminescence-activated multi-omic profiling. Together, these tools enable detailed mapping of the cellular biomolecular landscape. For example, CAP-seq revealed enrichment of transcripts encoding secretory and mitochondrial proteins near the endoplasmic reticulum membrane and outer mitochondrial membrane, supporting models of localized translation. Additionally, pulse-chase labeling using RinID in the ER lumen uncovered distinct decay kinetics of secretory proteins. Looking forward, future efforts may focus on developing low-toxicity and low-background chemical probes, engineering red-shifted photosensitizers for deep-tissue and in vivo applications, and integrating multiple proximity labeling (PL) platforms to study organelle contact sites and interorganelle molecular trafficking.
Fang, Y. and Zou, P.* (2025). Acc. Chem. Res. 58, 2526-2534.
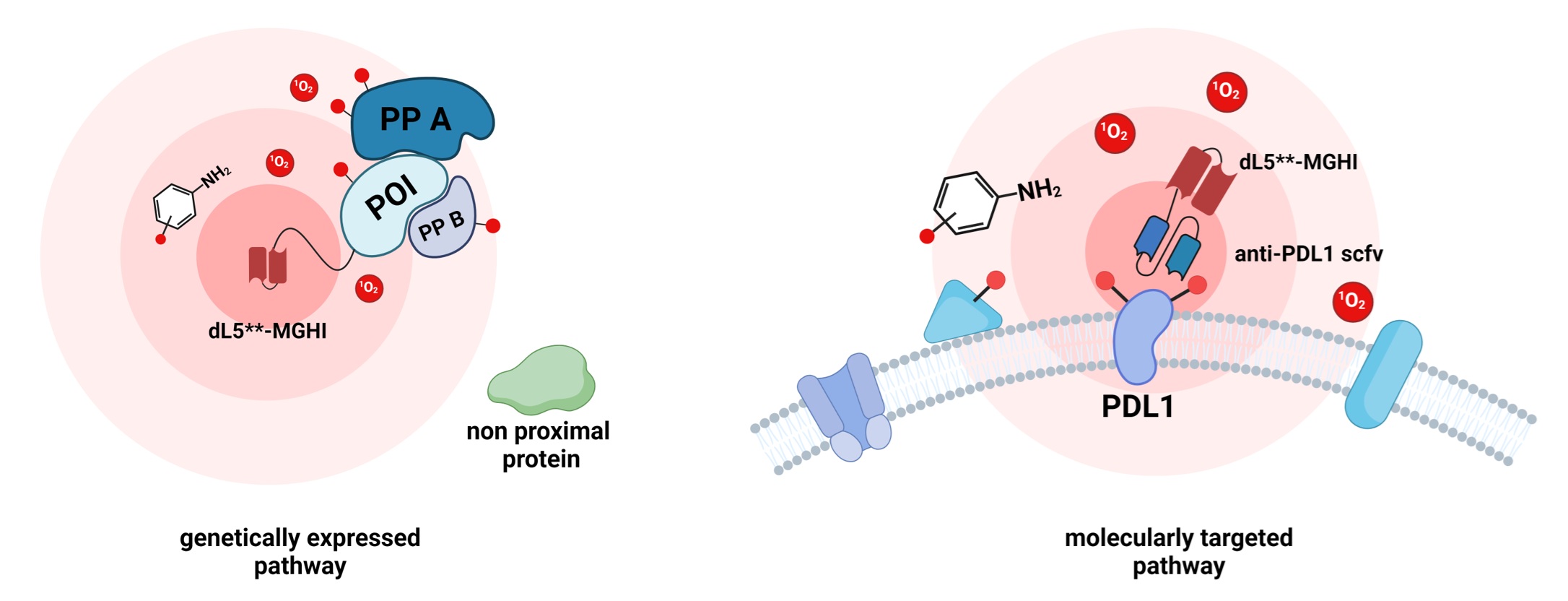
The spatial organization of proteins in eukaryotic cells plays essential roles in cellular functions. Genetically encoded proximity labeling methods offer spatially resolved and proteome-wide mapping of protein localization, yet existing techniques are limited to blue light activation, which has limited tissue penetration and causes a high cellular background. Here, we report the development of a near-infrared photocatalytic proximity labeling method, FLAPP, based on the engineered fluorogen-activating protein dL5**. Upon binding to the fluorogenic iodinated malachite green, the complex can efficiently absorb near-IR light to produce singlet oxygen that reacts in situ with nearby histidine residues. Unlike most existing near-infrared light-activated proximity labeling techniques that rely on antibody-dependent membrane targeting, FLAPP is a genetically encoded near-infrared light-activated proximity labeling technology. We demonstrate the high spatial specificity (96%) of FLAPP in the mitochondria and nucleus. FLAPP enables the deep tissue penetration of protein labeling, underscoring its potential for live animal applications.
Ren, T.#, Gong, J.#, Zheng, F., Long, J., Wang, H., He, J.*, Jiang, J.-H.* and Zou, P.* (2025). Anal. Chem. 97, 14492-14502.
73. Thioether editing generally increases the photostability of rhodamine dyes on self-labeling tags
Self-labeling protein tags are widely used in advanced bioimaging where dyes with high-photon budgets outperform their fluorescent protein counterparts. Further increasing the emitted photon numbers of dye-tag systems is actively pursued by both new fluorophore chemistry and protein engineering. By scrutinizing the protein microenvironment of fluorophores, here we propose that proximal thioether groups negatively affect the photostability of the dye-tag system. We attribute the disparity in photostability of rhodamine dyes on HaloTag, SNAP-tag, and TMP-tag3 to the influence of the inherent thioether linkage within the SNAP-tag and TMP-tag3. This photochemical pathway leads us to further devise tags with higher photostability. We first show that rhodamine dyes on TMP-tag3.1, which employs a proximity-induced SuFEx reaction instead of a thiol-acrylamide addition to replace the thioether adduct, achieve photon budgets comparable to those ligands on HaloTag. We further showcase that by mutating the methionine near the fluorophore pocket, HaloTag: M175L generally gives up to four times enhancement on photostability when labeled with red and far-red rhodamines. The enhancement of HaloTag modification is demonstrated with single-molecule fluorescence imaging, live-cell fluorescence imaging, and voltage imaging. During time-lapse imaging, gradual photooxidation of Met leads to a reduced photobleaching rate, mechanistically supporting the thioether pathway hypothesis. Our findings suggest that thioether editing on self-labeling tags is a general strategy to enhance the photostability of fluorophores for advanced time-lapse imaging techniques.
Ling, J.#, Zhang, Y.#, Hei, Y.#, Kompa, J., Yang, C., Wang, B., Zhang, J., Du, J., Rudi, T., Zhang, K., Sun, J., Wang, W., Fabritz, S., Li, Y., Deng, W., Zou, P., Chen, C.* and Chen, Z.* (2025). Proc. Natl. Acad. Sci. U. S. A. 122, e2426354122.
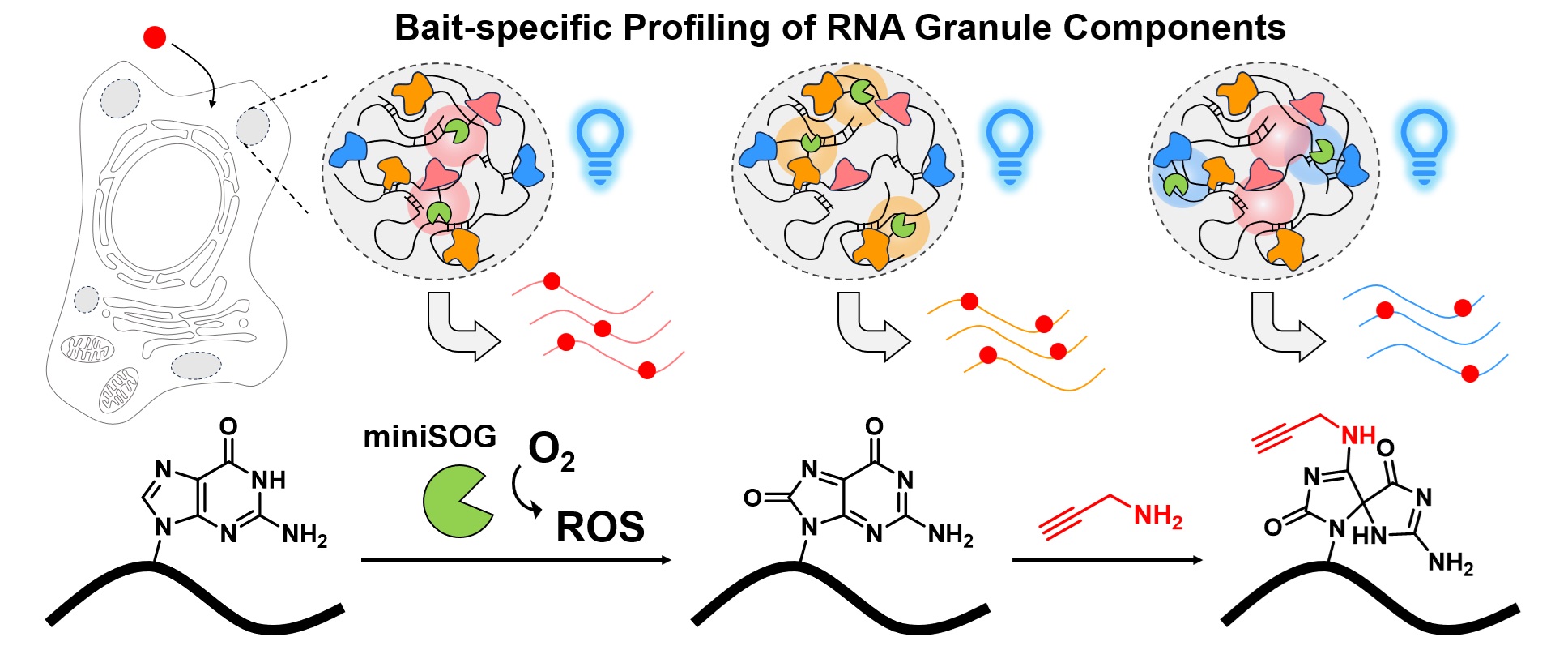
Stress granules (SGs) are dynamic, membrane-less organelles that house complex RNA-protein networks. Although previous profiling methods have characterized SG RNAs as long, translation-repressed, and extensively epigenetically modified, it remains unclear whether these RNAs are evenly distributed within SGs. In this study, we genetically targeted the photocatalyst protein miniSOG to multiple SG core proteins, enabling the comprehensive CAP-seq profiling of SG-associated RNAs. Our results reveal that RNAs near different SG core proteins display heterogeneous distributions and distinct intrinsic features. We also employed CAP-seq to map RNAs associated with processing body (PB) marker protein DDX6 under both unstressed conditions and arsenite-induced stress. By comparing the transcriptomes proximal to SGs and PBs, our data suggest that m6A modification may promote RNA localization to SGs, whereas higher AU content may facilitate mRNA targeting to PBs. These findings point to potential regulatory mechanisms that determine the subcellular localization of mRNAs within membrane-less organelles.
Ren, Z., Zhao, S., Tang, W. and Zou, P.* (2025). Anal. Chem. 97, 12767-12775.
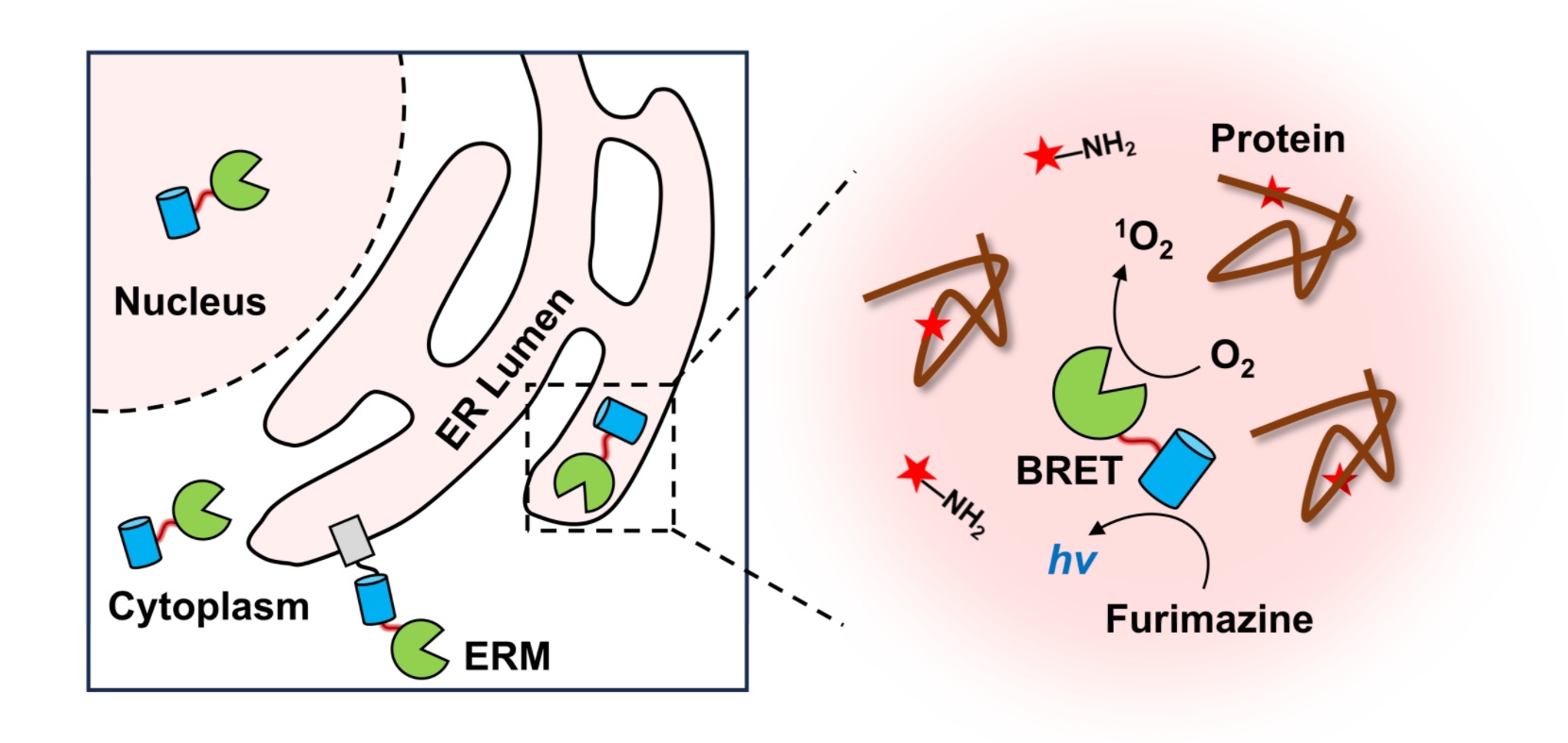
Mapping the spatial organization of proteins and cellular interactions is crucial for understanding their biological functions. Herein, we report a biocompatible, multi-functional luminescence-activated proximity labeling (LAP) strategy for profiling subcellular proteomes and cell-cell interactions in live cells and animals. Our method capitalizes on fusing the photocatalyst miniSOG to NanoLuc luciferase, whose bioluminescence activates miniSOG via a resonance energy transfer mechanism, generating reactive oxygen species in situ to mediate proximity labeling. We demonstrated the high spatial specificity of LAP in a C57BL6/N mouse model transplanted with MC38 cells. Our data revealed tumor microenvironment-dependent remodeling of secretome. LAP was further applied to identify ligand-receptor mediated cell-cell interactions both in vitro and in vivo. We also achieved local transcriptome profiling by combining LAP with next-generation sequencing. Overall, LAP was proved to be a versatile proximity labeling technique with strong biocompatibility for spatial multi-omic applications.
Wang, R.#, Fang, Y.#, Hu, Y., Liu, Y.*, Chen, P. R.*, Zou, P.* (2025). Chem 11, 102595.
Biological fluorescence imaging is constrained by a trade-off between field of view (FoV) and acquisition rate. This limitation is particularly evident with scientific complementary metal oxide semiconductor (sCMOS) cameras, where high frame rates force the maximum FoV into a narrow, high aspect ratio rectangle. Moreover, the rolling shutter used in high-speed imaging produces non-uniform exposure across the FoV, introducing artifacts when capturing rapid dynamics such as fast-moving objects or fluctuating fluorescence signals. These issues are especially detrimental to voltage imaging, a key technique for studying nervous system dynamics. Here, we introduce ROSE-based high-speed imaging (ROSE-HSI), a method that increases pixel throughput sixfold (when compared with a square FoV) by selectively exposing two cameras. ROSE-HSI not only overcomes the sampling rate limitations of a single camera but also eliminates the temporal inaccuracies caused by the rolling shutter. Our technique has enabled simultaneous kilohertz voltage imaging over a 700x732-pixel area, capturing distinct neuronal waveforms and synaptic-mediated signal propagation with high fidelity.
Xiong, S.#, Peng, L.#, Gu, L.*, Zou, P.* and Ji, W.* (2025). Optica 12, 860-871.
Organic fluorophores are the keystone of advanced biological imaging. The vast chemical space of fluorophores has been extensively explored in search of molecules with ideal properties. However, within the current molecular constraints, there appears to be a trade-off between high brightness, robust photostability, and tunable biochemical properties. Herein we report a general strategy to systematically boost the performance of donor-acceptor-type fluorophores, such as rhodamines, by leveraging SO2 and O-substituted azabicyclo[3.2.1] octane auxochromes. These bicyclic heterocycles give rise to a collection of ‘bridged’ dyes (BD) spanning the ultraviolet and visible range with top-notch quantum efficiencies, enhanced water solubility, and tunable cell-permeability. Notably, these azabicyclic fluorophores showed remarkable photostability compared to their tetramethyl or azetidine analogs while being completely resistant to oxidative photoblueing. Functionalized BD dyes are tailored for applications in single-molecule imaging, super-resolution imaging (STED and SIM) in fixed or live mammalian and plant cells, and live zebrafish imaging and chemogenetic voltage imaging.
Zhang, J., Zhang, K., Wang, K., Wang, B., Zhu, S., Qian, H., Ma, Y., Zhang, M., Liu, T., Chen, P., Shen, Y., Fu, Y., Fang, S., Zhang, X., Zou, P., Deng, W., Mu, Y., Chen, Z.* (2025). Nat. Methods 22, 1276-1287.
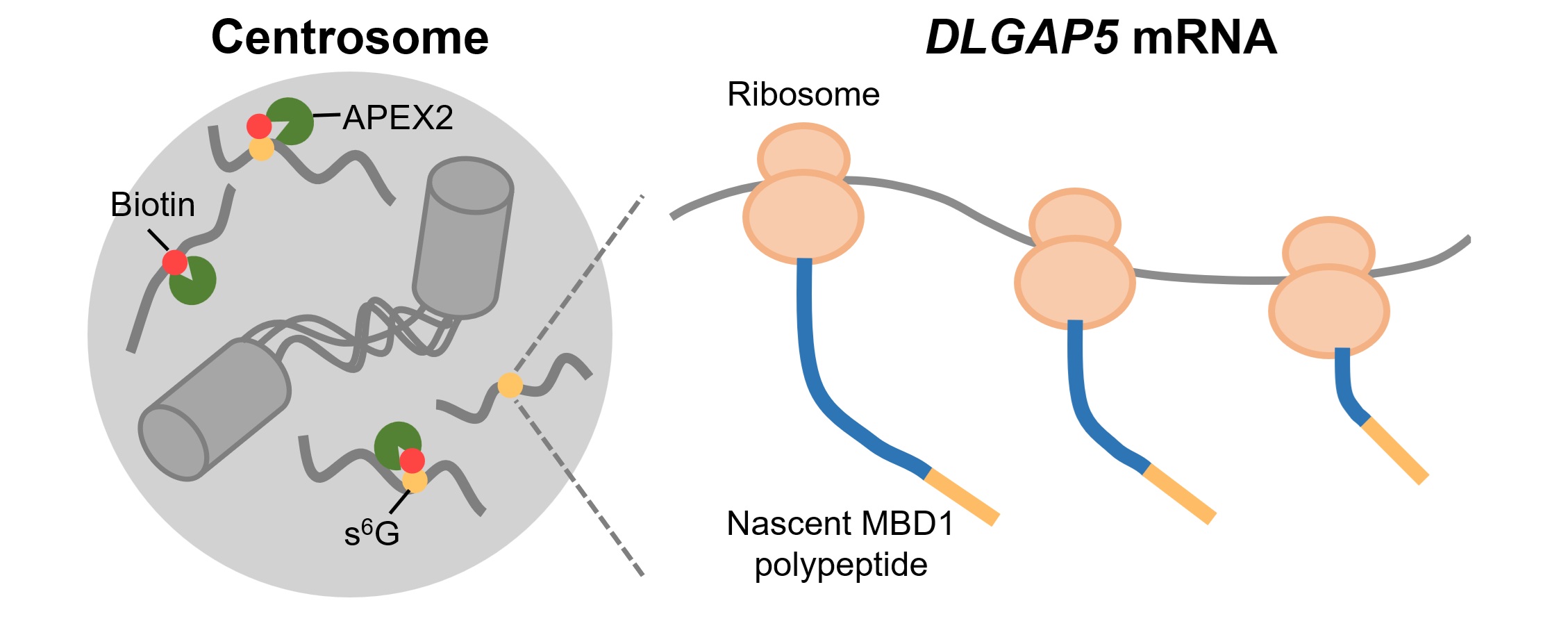
Subcellular RNA localization is a conserved mechanism in eukaryotic cells and plays critical roles in diverse physiological processes including cell proliferation, differentiation, and embryo development. Nevertheless, the characterization of centrosome-localized mRNAs remains under-explored due to technical difficulties. In this study, we utilize APEX2-mediated proximity labeling to map centrosome-proximal transcriptome, identifying DLGAP5 mRNA as a novel centrosome-localized transcript during mitosis. Using a combination of drug perturbation, truncation, deletion, and mutagenesis, we demonstrate that microtubule binding of nascent MBD1 polypeptide is required for centrosomal transport of DLGAP5 mRNA. Our data also reveals that mRNA targeting efficiency is tightly linked to the coding sequence (CDS) length. Thus, our study provides a transcriptomic resource for future investigation of centrosome-localized RNAs, and sheds light on mechanisms underlying mRNA centrosomal localization.
Wang, G., Li, M.* and Zou, P.* (2025). RSC Chem. Biol. 6, 919-932.
Qu, D., Li, Y., Liu, Q., Cao, B., Cao, M., Lin, X., Shen, C., Zou, P., Zhou, H.*, Zhang, W.* and Pan, W.* (2025). Cell Res. 35, 149-152.
66. Recent development of chemigenetic hybrid voltage indicators enabled by bioconjugation chemistry
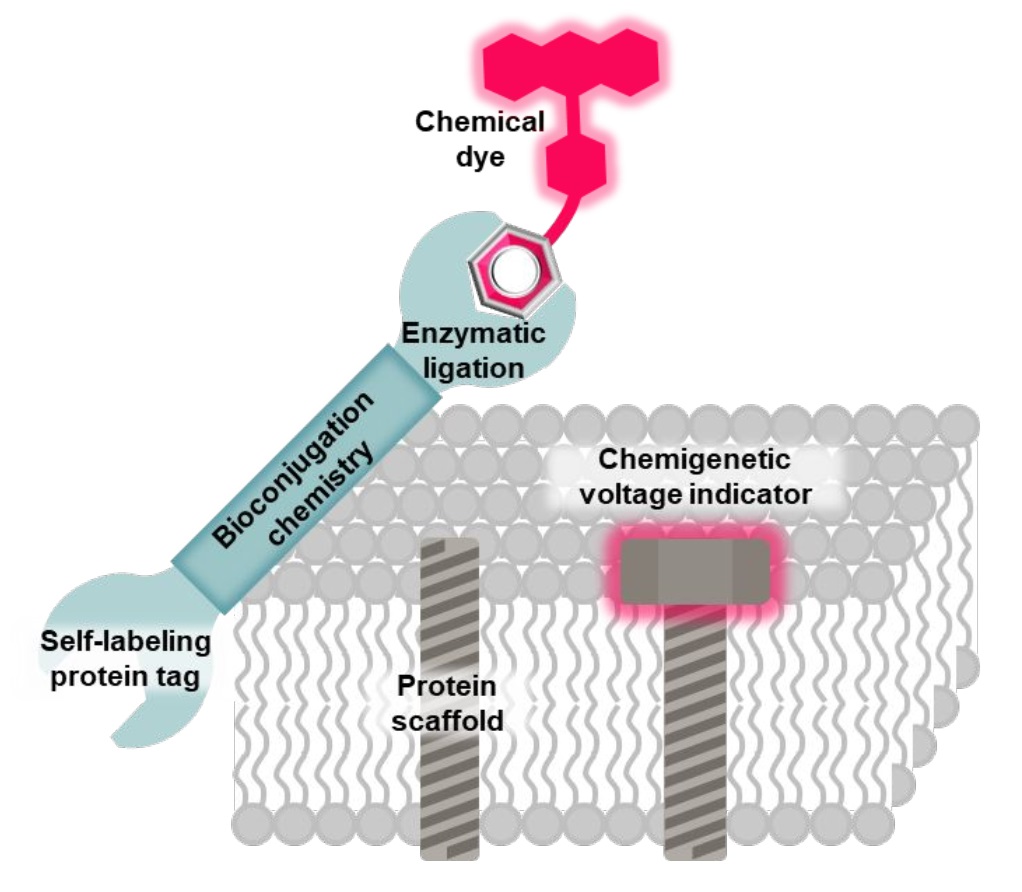
Fluorescent voltage indicators enable the optical recording of electrophysiology across large cell populations with subcellular resolution; however, their application is often constrained by a limited photon budget. To address this limitation, advanced bioconjugation methods have been employed to site-specifically attach bright and photostable organic dyes to cell-specific protein scaffolds in live cells. The resulting chemigenetic hybrid voltage indicators enable sustained monitoring of voltage fluctuations with an exceptional signal-to-noise ratio, both in vitro and in vivo. This Viewpoint discusses recent advancements in the development of these indicators through bioconjugation chemistry.
Liu, S. and Zou, P.* (2024). Bioconjug. Chem. 35, 1711.
Rhodamines have been continuously optimized in brightness, biocompatibility, and color to fulfill the demands of modern bioimaging. However, the problem of phototoxicity caused by the excited fluorophore under long-term illumination has been largely neglected, hampering their use in time-lapse imaging. Here we introduce cyclooctatetraene (COT) conjugated rhodamines that span the visible spectrum and exhibit significantly reduced phototoxicity. We identified a general strategy for the generation of Gentle Rhodamines, which preserved their outstanding spectroscopic properties and cell permeability while showing an efficient reduction of singlet-oxygen formation and diminished cellular photodamage. Paradoxically, their photobleaching kinetics do not go hand in hand with reduced phototoxicity. By combining COT-conjugated spirocyclization motifs with targeting moieties, these Gentle Rhodamines compose a toolkit for time-lapse imaging of mitochondria, DNA, and actin, and synergize with covalent and exchangeable HaloTag labeling of cellular proteins with less photodamage than their commonly used precursors. Taken together, the Gentle Rhodamines generally offer alleviated phototoxicity and allow advanced video recording applications, including voltage imaging.
Liu, T., Kompa, J., Ling, J., Lardon, N., Zhang, Y., Chen, J., Reymond, L., Chen, P., Tran, M., Yang, Z., Zhang, H., Liu, Y., Pitsch, S., Zou, P., Wang, L., Johnsson, K. and Chen, Z.* (2024). ACS Cent. Sci. 10, 1933.
The development of new or improved single fluorescent protein (FP)-based biosensors (SFPBs), particularly those with excitation and emission at near-infrared wavelengths, is important for the continued advancement of biological imaging applications. In an effort to accelerate the development of new SFPBs, we report modified transposons for the transposase-based creation of libraries of FPs randomly inserted into analyte binding domains, or vice versa. These modified transposons feature ends that are optimized to minimize the length of the linkers that connect the FP to the analyte binding domain. We rationalized that shorter linkers between the domains should result in more effective allosteric coupling between the analyte binding-dependent conformational change in the binding domain and the fluorescence modulation of the chromophore of the FP domain. As a proof of concept, we employed end-modified Mu transposons for the discovery of SFPB prototypes based on the insertion of two circularly permuted red FPs (mApple and FusionRed) into binding proteins for l-lactate and spermidine. Using an analogous approach, we discovered calcium ion (Ca2+)-specific SFPBs by random insertion of calmodulin (CaM)-RS20 into miRFP680, a particularly bright near-infrared (NIR) FP based on a biliverdin (BV)-binding fluorescent protein. Starting from an miRFP680-based Ca2+ biosensor prototype, we performed extensive directed evolution, including under BV-deficient conditions, to create highly optimized biosensors designated the NIR-GECO3 series. We have extensively characterized the NIR-GECO3 series and explored their utility for biological Ca2+ imaging. The methods described in this work will serve to accelerate SFPB development and open avenues for further exploration and optimization of SFPBs across a spectrum of biological applications.
Chai, F., Fujii, H., Le, G. N. T., Lin, C., Ota, K., Lin, K. M., Pham, L. M. T., Zou, P., Drobizhev, M., Nasu, Y., Terai, T., Bito, H. and Campbell, R. E.* (2024). ACS Sens. 9, 3394.
Bioorthogonal chemistry was deservedly recognized with the 2022 Nobel Prize in Chemistry, having transformed the way chemists and biologists interrogate biological systems in the past twenty years. This Voices piece asks researchers from a range of backgrounds: what are some major challenges and opportunities facing the field in coming years?
Hsu, K.-L., Schumann, B., Sletten, E., Vinogradova, E. and Zou, P. (2024). Cell Chem. Biol. 31, 380.
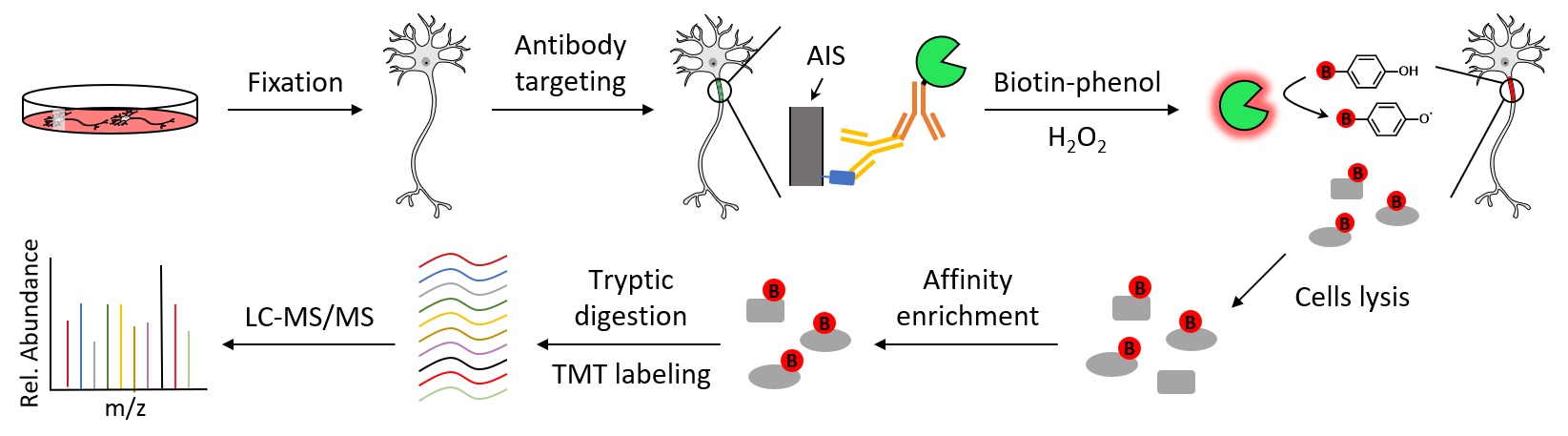
The axon initial segment (AIS) is a specialized neuronal compartment required for action potential generation and neuronal polarity. However, understanding the mechanisms regulating AIS structure and function has been hindered by an incomplete knowledge of its molecular composition. Here, using immuno-proximity biotinylation we further define the AIS proteome and its dynamic changes during neuronal maturation. Among the many AIS proteins identified, we show that SCRIB is highly enriched in the AIS both in vitro and in vivo, and exhibits a periodic architecture like the axonal spectrin-based cytoskeleton. We find that ankyrinG interacts with and recruits SCRIB to the AIS. However, loss of SCRIB has no effect on ankyrinG. This powerful and flexible approach further defines the AIS proteome and provides a rich resource to elucidate the mechanisms regulating AIS structure and function.
Zhang, W., Fu, Y., Peng, L., Ogawa, Y., Ding, X., Rasband, A., Zhou, X., Shelly, M., Rasband M. N.* and Zou, P.* (2023). Nat. Commun. 14, 8201.

Genetically encoded fluorescent indicators (GEVIs) allow the direct visualization of cellular membrane potential at the millisecond time scale. Among these, red-emitting GEVIs have been reported to support multi-channel recordings and manipulation of cellular activities with reduced auto-fluorescence background. However, the limited sensitivity and dimness of existing red GEVIs have restricted their applications in neuroscience. Here, we report a pair of red-shifted opsin-based GEVIs, Cepheid1b and Cepheid1s, with improved dynamic range, brightness, and photostability. The improved dynamic range is achieved by a rational design to raise the electrochromic Förster resonance energy transfer (eFRET) efficiency, and the higher brightness and photostability is approached with separately engineered red fluorescent proteins. With Cepheid1 indicators, we recorded complex firings as well as subthreshold activities of neurons on acute brain slices, and observed heterogeneity in the voltage-calcium-coupling on pancreatic islets. Overall, Cepheid1 indicators provide a strong tool to investigate excitable cells in various sophisticated biological systems.
Han, Y.#, Yang, J.#,*, Li, Y.#, Chen, Y.#, Ren, H., Ding, R., Qian, W., Ren, K., Xie, B., Deng, M., Xiao, Y., Chu, J. and Zou, P.* (2023) Sci. Adv. 9, eadi4208.
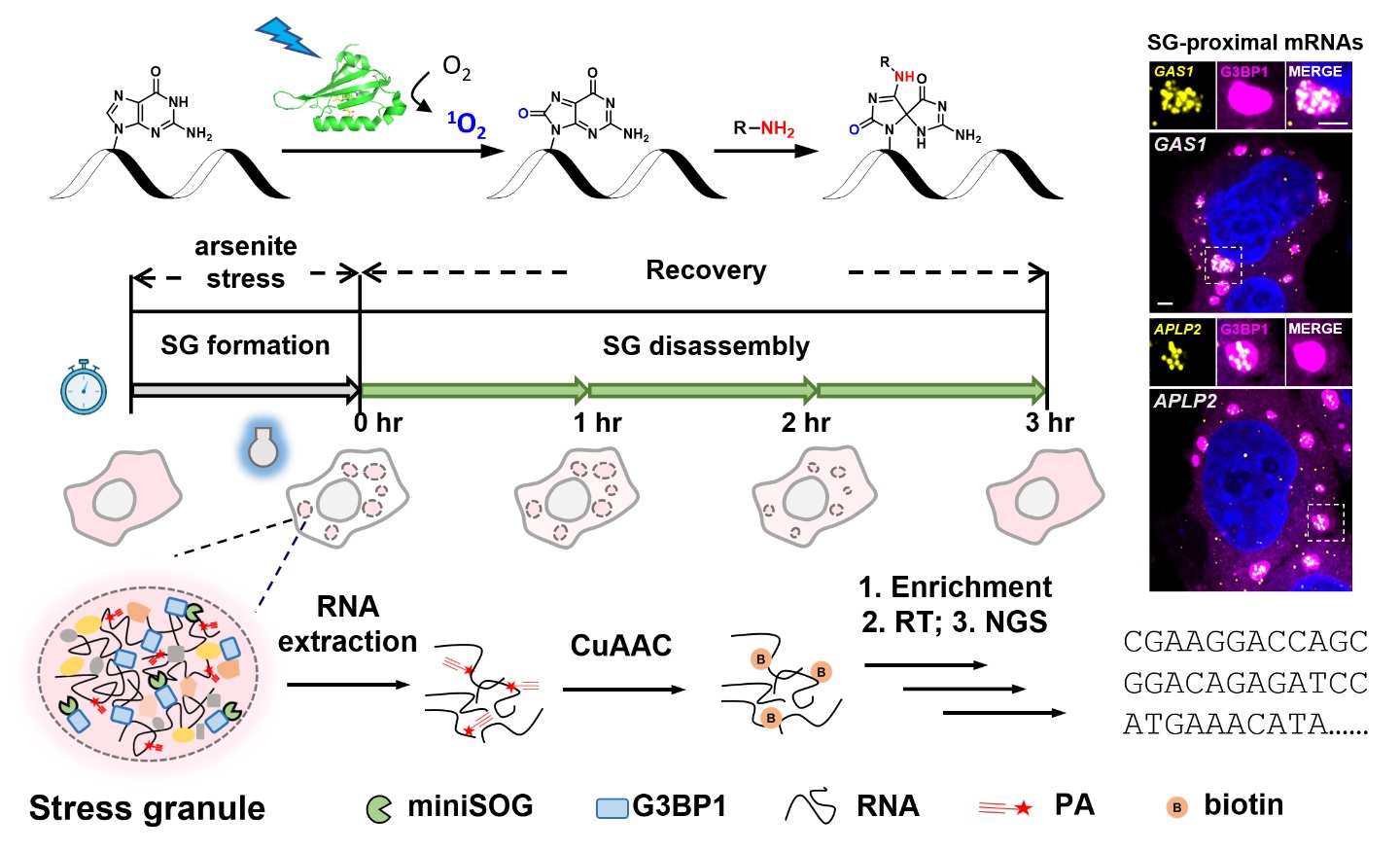
Stress granules (SGs) are highly dynamic cytoplasmic membrane-less organelles that assemble when cells are challenged by stress. RNA molecules are sorted into SGs where they play important roles in maintaining the structural stability of SGs and regulating gene expression. Herein, we apply a proximity-dependent RNA labeling method, CAP-seq, to comprehensively investigate the content of SG-proximal transcriptome in live mammalian cells. CAP-seq captures 457 and 822 RNAs in arsenite- and sorbitol-induced SGs in HEK293T cells, respectively, revealing that SG enrichment is positively correlated with RNA length and AU content, but negatively correlated with translation efficiency. The high spatial specificity of CAP-seq dataset is validated by single-molecule FISH imaging. We further apply CAP-seq to map dynamic changes in SG-proximal transcriptome along the time course of granule assembly and disassembly processes. Our data portray a model of AU-rich and translationally repressed SG nanostructure that are memorized long after the removal of stress.
Ren, Z.#, Tang, W.#, Peng, L. and Zou, P.* (2023) Nat. Commun. 14, 7390.
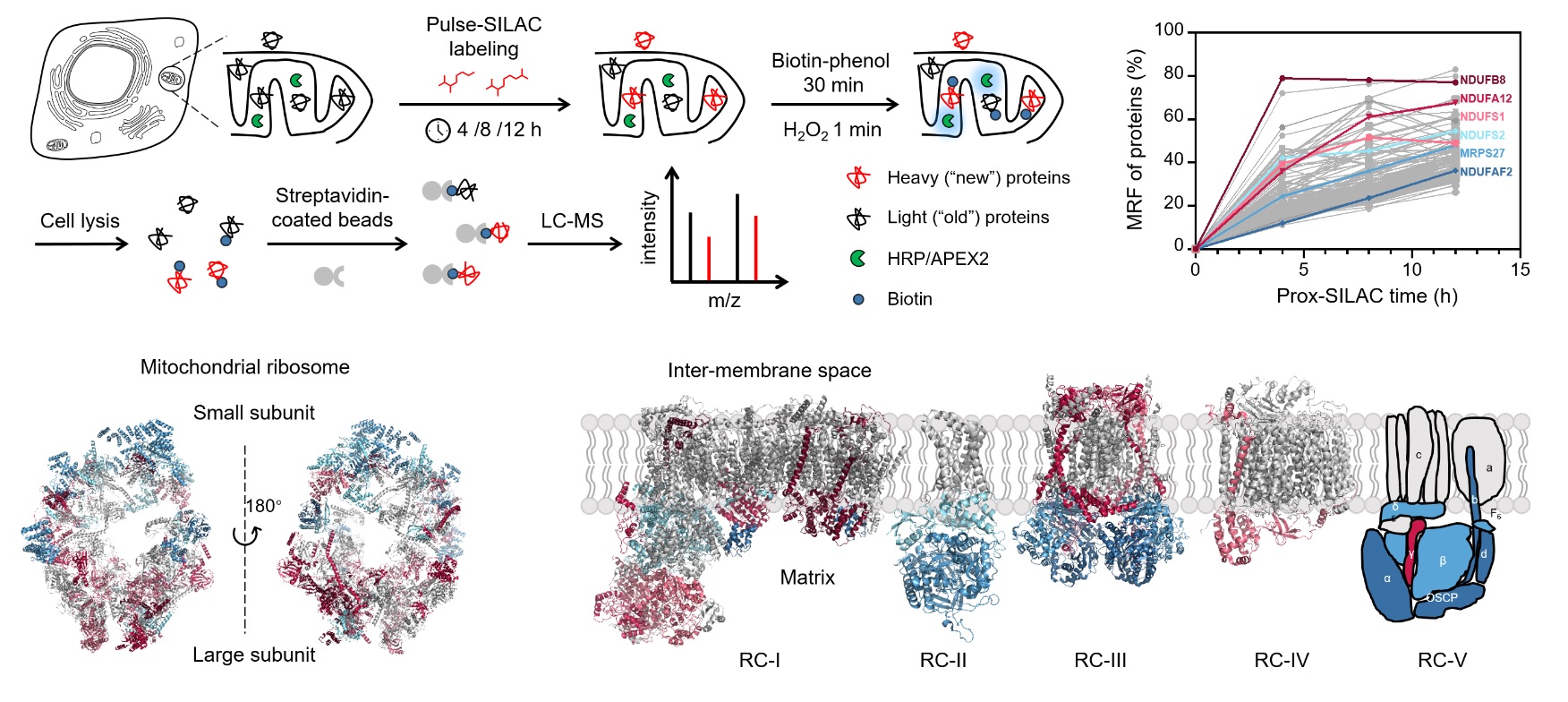
Cellular activities are commonly associated with dynamic proteomic changes at the subcellular level. Although several techniques are available to quantify whole-cell protein turnover dynamics, such measurements often lack sufficient spatial resolution at the subcellular level. Herein, we report the development of prox-SILAC method that combines proximity-dependent protein labeling (APEX2/HRP) with metabolic incorporation of stable isotopes (pulse-SILAC) to map newly synthesized proteins with subcellular spatial resolution. We apply prox-SILAC to investigate proteome dynamics in the mitochondrial matrix and the endoplasmic reticulum (ER) lumen. Our analysis reveals a highly heterogeneous distribution in protein turnover dynamics within macromolecular machineries such as the mitochondrial ribosome and respiratory complexes I-V, thus shedding light on their mechanism of hierarchical assembly. Furthermore, we investigate the dynamic changes of ER proteome when cells are challenged with stress or undergoing stimulated differentiation, identifying subsets of proteins with unique patterns of turnover dynamics, which may play key regulatory roles in alleviating stress or promoting differentiation. We envision that prox-SILAC could be broadly applied to profile protein turnover at various subcellular compartments, under both physiological and pathological conditions.
Yuan, F.#, Li, Y.#, Zhou, X. and Zou, P.* (2023) Nat. Commun. 14, 7217.
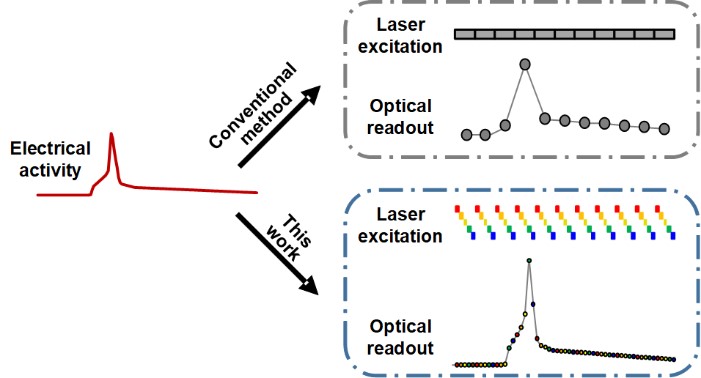
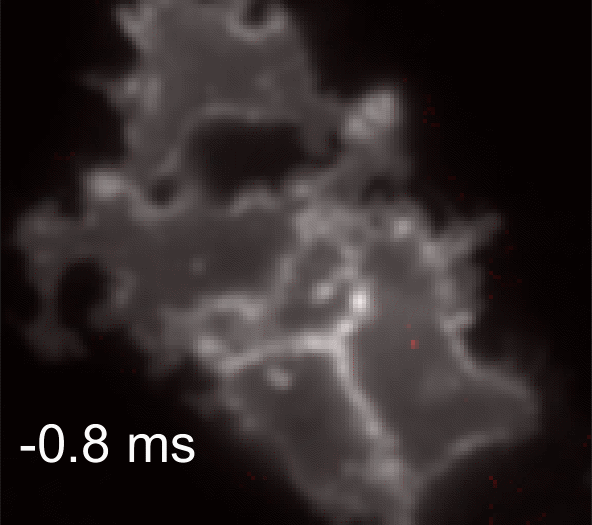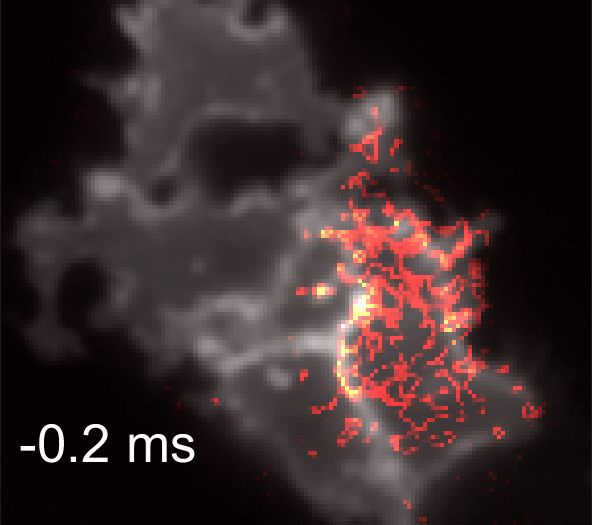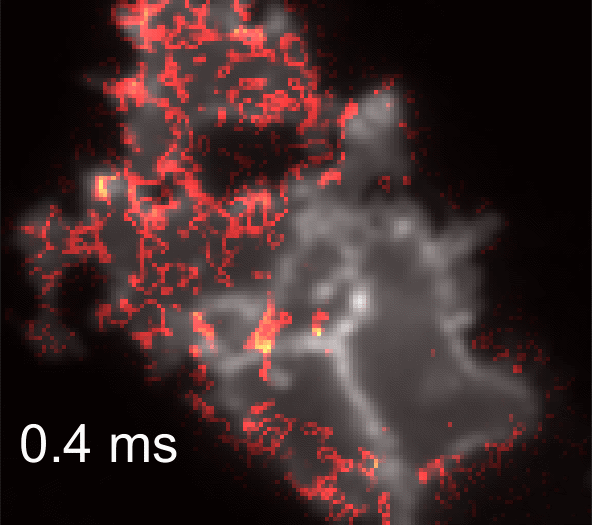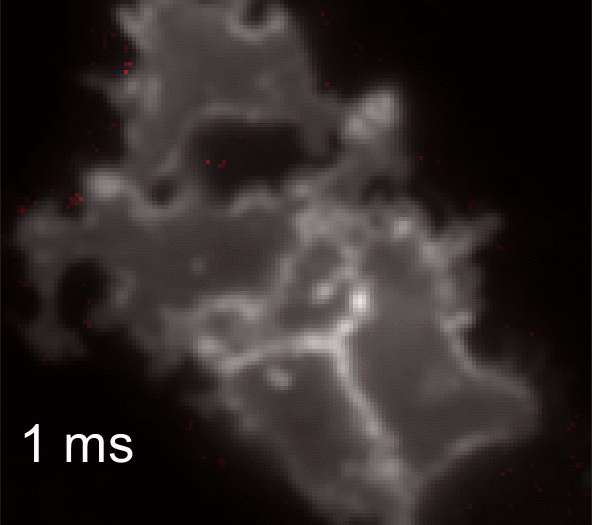
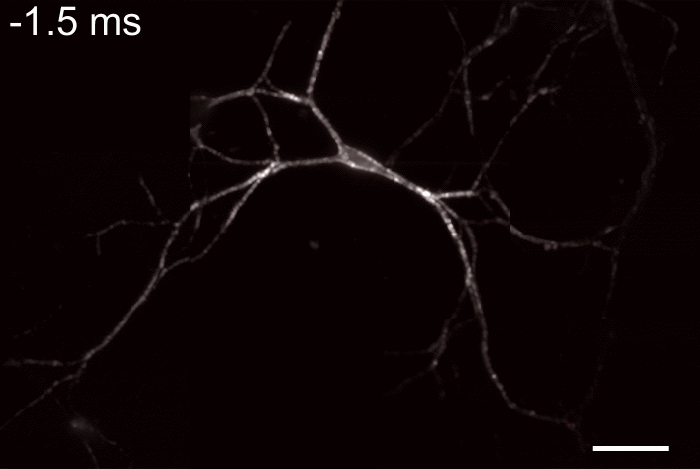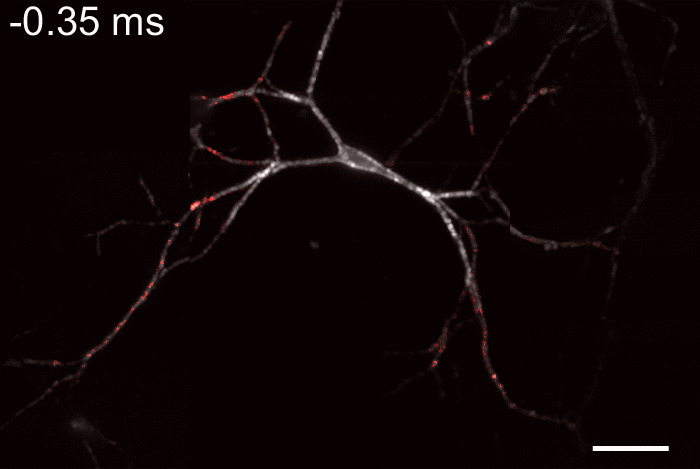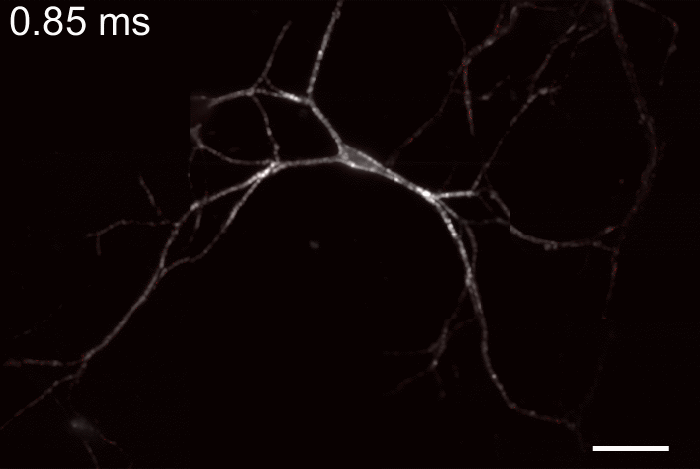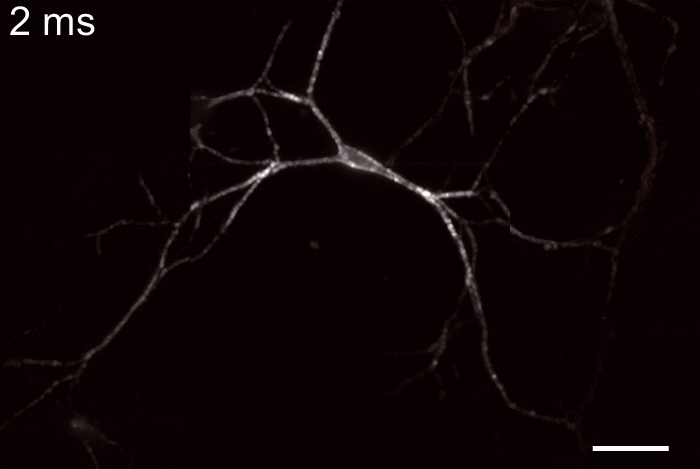
Membrane potential and its fluctuation are fundamental biophysical phenomena essential to cellular activities and functions. Compared to traditional electrode-based techniques, the optical recording via developed genetically-encoded voltage indicators (GEVIs) offers a combination of non-invasiveness, high spatial resolution, and increased measurement throughput. However, its application is limited by the insufficient acquisition rate and time accuracy of the camera. Here we design and apply a stroboscopic illumination scheme to boost the temporal resolution of voltage imaging, while simultaneously eliminating the artifacts caused by non-synchronized exposure in the rolling-shutter mode. We demonstrate that commonly used GEVIs are compatible with stroboscopic voltage imaging (SVI), and our SVI scheme offers a 5-fold faster acquisition frame rate than conventional continuous illumination. The GEVIs tested maintain high sensitivities in the SVI mode, supporting faithful reports of intracellular depolarization waveform and intercellular gap junction-mediated depolarization coupling in human embryonic kidney 293T (HEK 293T) cell populations. SVI allows resolving the action potential (AP) waveform with less distortion and mapping action potential initiation and propagation dynamics in cultured neurons in kilohertz, beyond the restriction from the camera in the field of view.
Peng, L. and Zou, P.* (2023) Chem. Biomed. Imaging 1, 448–460.
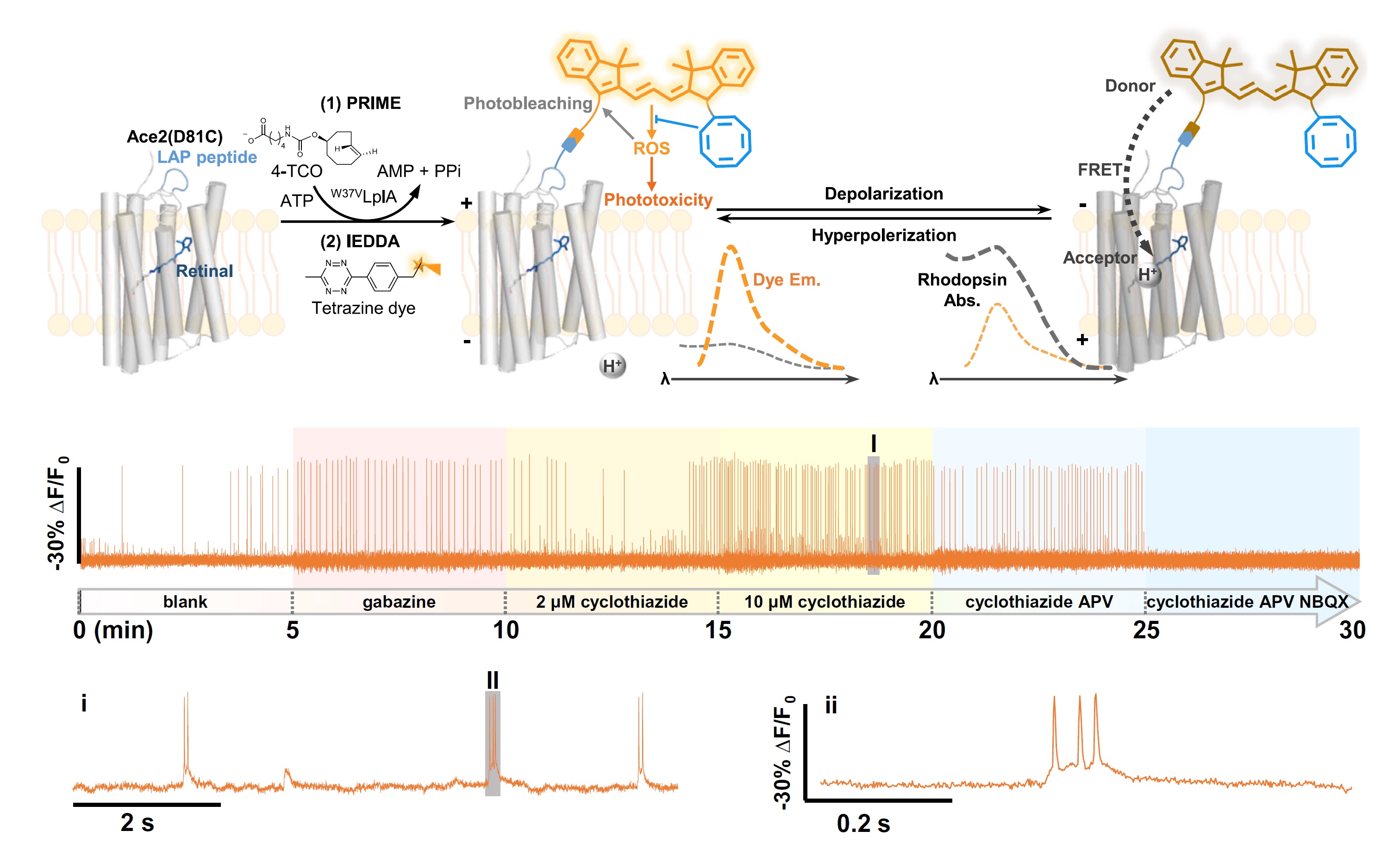
HVI is a chemo-genetic sensor that combines the superior photophysical properties of organic dyes and the genetic targetability of protein sensors to report transient membrane voltage changes. It exhibits boosted sensitivity in excitable cells such as neurons and cardiomyocytes. However, the voltage signals recorded during long-term imaging are severely diminished or distorted due to phototoxicity and photobleaching issues. To capture stable electrophysiological activities over a long time, we employ cyanine dyes conjugated with a COT molecule as the fluorescence reporter of HVI. The resulting orange-emitting HVI-COT-Cy3 enables high-fidelity voltage imaging for up to 30 min in cultured primary neurons with a sensitivity of ~-30% ΔF/F0 per action potential (AP). It also maximally preserves the signal of individual APs in cardiomyocytes. The far-red-emitting HVI-COT-Cy5 allows two-color voltage/calcium imaging with GCaMP6s in neurons and cardiomyocytes for 15 min. We leverage the HVI-COT series with reduced phototoxicity and photobleaching to evaluate the impact of drug candidates on the electrophysiology of excitable cells.
Liu, S.#, Ling, J.#, Chen, P., Cao, C., Peng, L., Zhang, Y., Ji, G., Guo, Y., Chen, P. R., Zou, P.* and Chen, Z.* (2023) Proc. Natl. Acad. Sci. U. S. A. 120, e2306950120.
56. Functional imaging-guided cell selection for evolving genetically encoded fluorescent indicators
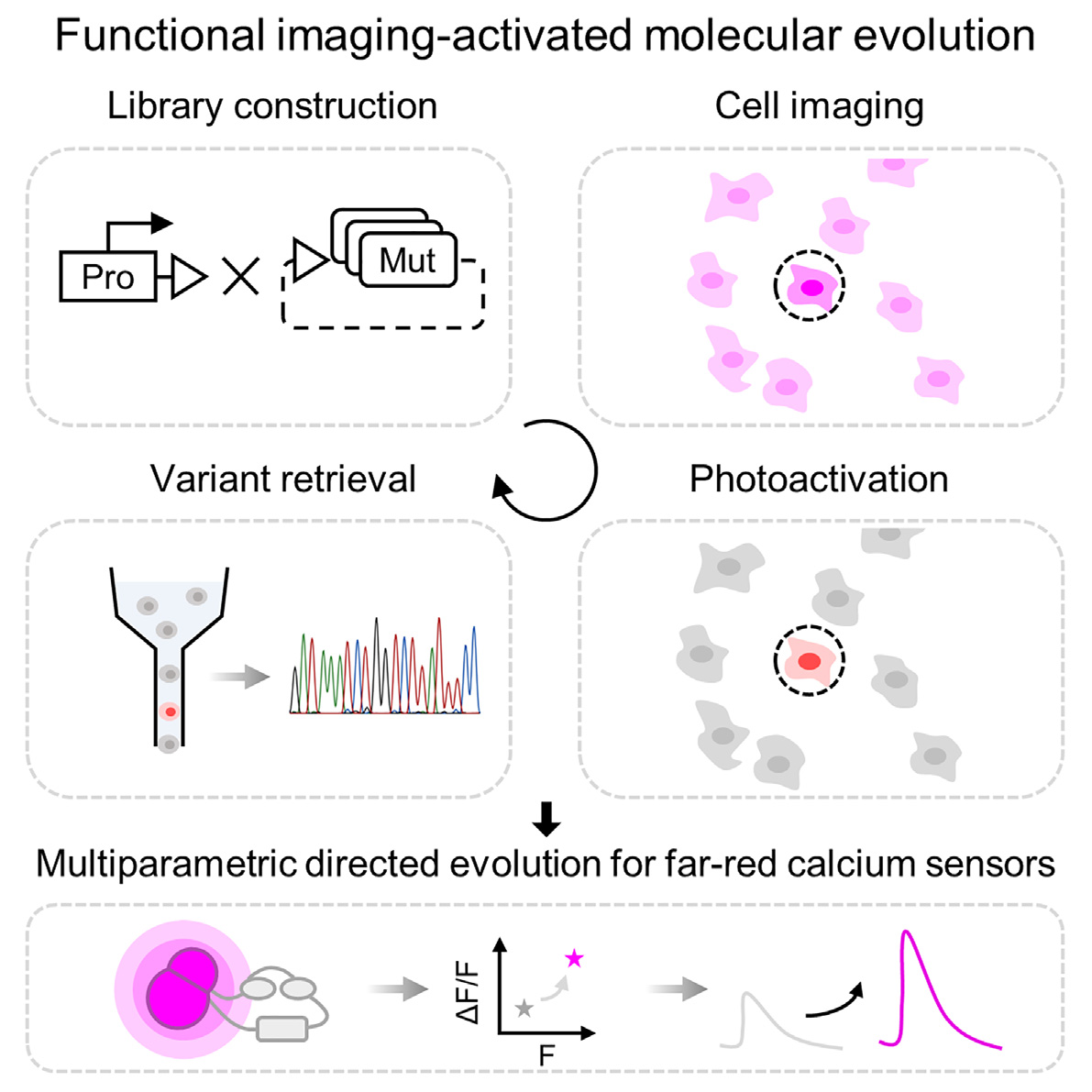
Genetically encoded fluorescent indicators are powerful tools for tracking cellular dynamic processes. Engineering these indicators requires balancing screening dimensions with screening throughput. Herein, we present a functional imaging-guided photoactivatable cell selection platform, Faculae, for linking microscopic phenotype with the underlying genotype in a pooled mutant library. Faculae is capable of assessing tens of thousands of variants in mammalian cells simultaneously while achieving photoactivation with single cell resolution in seconds. To demonstrate the feasibility of this approach, we applied Faculae to perform multidimensional directed evolution for far-red genetically encoded calcium indicators (FR-GECIs) with improved brightness (Nier1b) and SBR (Nier1s). We anticipate that this image-based pooled screening method will facilitate the development of a wide variety of biomolecular tools.
Lin, C., Liu, L. and Zou, P.* (2023) Cell Rep. Methods 3, 100544.
In eukaryotic cells, the subcellular targeting of RNA controls many fundamental aspects of cellular physiology. Despite broad distributions throughout the cytoplasm, RNA molecules are conventionally believed to be excluded from the secretory pathway compartments including the endoplasmic reticulum (ER). Recent discovery of RNA N-glycan modification (glycoRNAs) has challenged this view, but direct evidence of RNA localization in the ER lumen has been lacking. In this study, we applied enzyme-mediated proximity labeling to profile the ER lumen-localized RNAs in human embryonic kidney 293T cells and rat cortical neurons. Our data set reveals the presence of small non-coding RNAs in the ER lumen, including U RNAs and Y RNAs, which raises interesting questions regarding their transport mechanism and biological functions in the ER.
Ren, Z.#, Li, R.#, Zhou, X.#, Chen, Y., Fang, Y. and Zou, P.* (2023) Biochemistry 62, 1844.
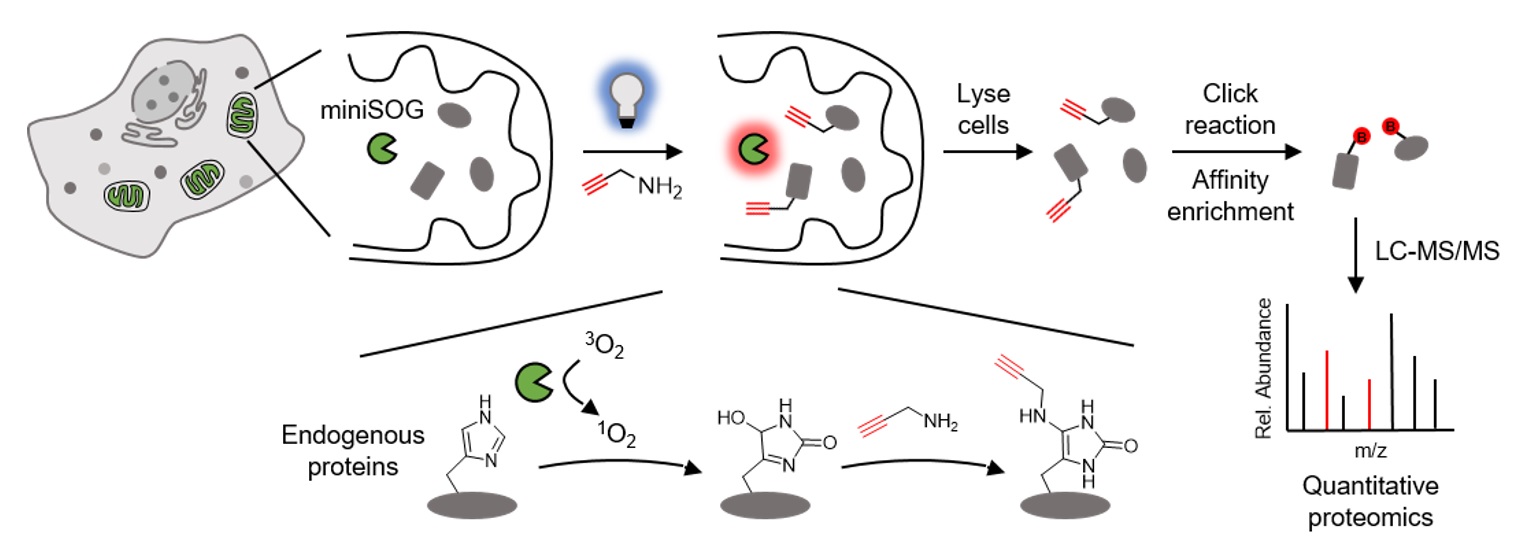
Mapping the subcellular organization of proteins is crucial for understanding their biological functions. Herein, we report a reactive oxygen species induced protein labeling and identification (RinID) method for profiling subcellular proteome in the context of living cells. Our method capitalizes on a genetically encoded photocatalyst, miniSOG, to locally generate singlet oxygen that reacts with proximal proteins. Labeled proteins are conjugated in situ with an exogenously supplied nucleophilic probe, which serves as a functional handle for subsequent affinity enrichment and mass spectrometry-based protein identification. From a panel of nucleophilic compounds, we identify biotin-conjugated aniline and propargyl amine as highly reactive probes. As a demonstration of the spatial specificity and depth of coverage in mammalian cells, we apply RinID in the mitochondrial matrix, capturing 477 mitochondrial proteins with 94% specificity. We further demonstrate the broad applicability of RinID in various subcellular compartments, including the nucleus and the endoplasmic reticulum (ER). The temporal control of RinID enables pulse-chase labeling of ER proteome in HeLa cells, which reveals substantially higher clearance rate for secreted proteins than ER resident proteins.
Zheng, F., Yu, C., Zhou, X. and Zou, P.* (2023) Nat. Commun. 14, 2978.
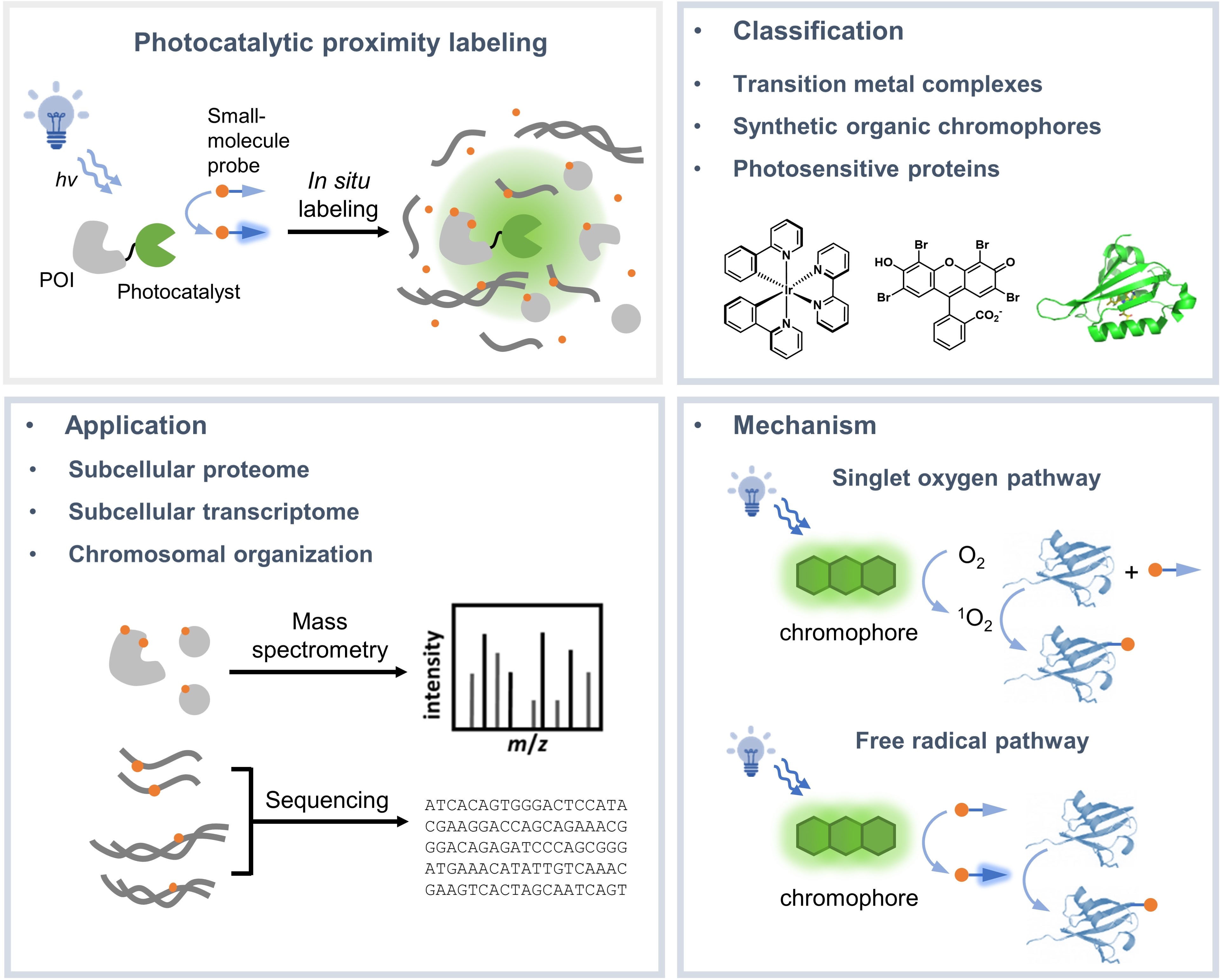
Investigating the subcellular organization of biomolecules is important for understanding their biological functions. Over the past decade, proximity-dependent labeling methods have emerged as powerful tools for mapping biomolecules in their native context. These methods often capitalize on the in-situ generation of highly reactive intermediates for covalently tagging biomolecules located within nanometers to sub-micrometers of the source of labeling. Among these, photocatalytic proximity labeling methods achieve precise spatial and temporal control of labeling with visible light illumination. In this review, we summarize the mechanisms and applications of existing photocatalytic proximity labeling methods and discuss future opportunities for improving the method.
Fang, Y. and Zou, P.* (2023) Chembiochem. 24, e202200745.
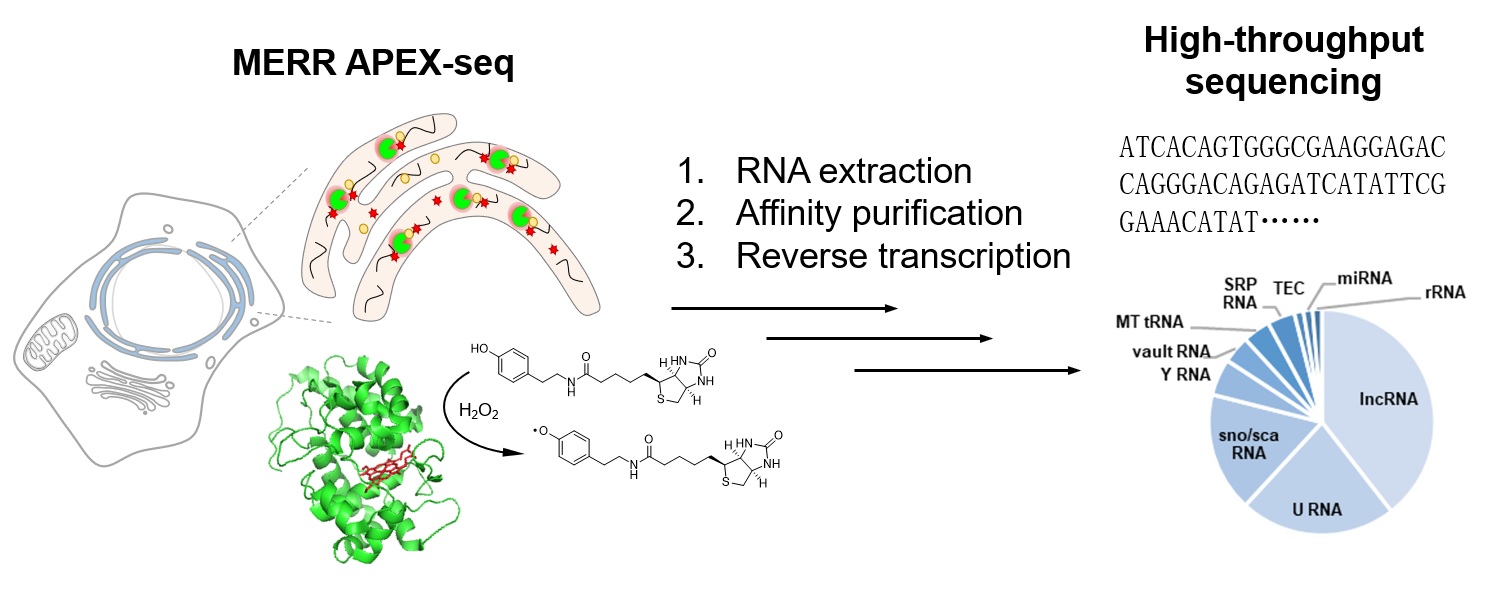
Knowledge about the spatial organization of RNAs in eukaryotic cells is crucial for understanding their functions. Here, we present a detailed MERR APEX-seq protocol to achieve spatiotemporally resolved mapping of the subcellular transcriptome in cultured mammalian cells. This protocol provides detailed description of constructing cell lines stably expressing APEX2, immunofluorescence characterization, MERR APEX labeling, enrichment of biotinylated RNA, library construction and high-throughput sequencing, and MERR APEX-seq data analysis. For complete details on the use and execution of this protocol, please refer to Li et al. (2022).
Li, R. and Zou, P.* (2023) STAR Protoc. 4, 102057.
Wang, R. and Zou, P.* (2023) ACS Cent. Sci. 9, 349-351.
Bernardes, G. J. L., Zou, P., Dai, Z., Lavik, E., van Hest, J., Zheng, G., Quinn, N., MacLaughlin, C. M., Reineke, T. M.* (2023) Bioconjugate Chem. 34, 279–282.
The differentiation of pluripotent stem cells (PSCs) into diverse functional cell types provides a promising solution to support drug discovery, disease modeling, and regenerative medicine. However, functional cell differentiation is currently limited by the substantial line-to-line and batch-to-batch variabilities, which severely impede the progress of scientific research and the manufacturing of cell products. For instance, PSC-to-cardiomyocyte (CM) differentiation is vulnerable to inappropriate doses of CHIR99021 (CHIR) that are applied in the initial stage of mesoderm differentiation. Here, by harnessing live-cell bright-field imaging and machine learning (ML), we realize real-time cell recognition in the entire differentiation process, e.g., CMs, cardiac progenitor cells (CPCs), PSC clones, and even misdifferentiated cells. This enables non-invasive prediction of differentiation efficiency, purification of ML-recognized CMs and CPCs for reducing cell contamination, early assessment of the CHIR dose for correcting the misdifferentiation trajectory, and evaluation of initial PSC colonies for controlling the start point of differentiation, all of which provide a more invulnerable differentiation method with resistance to variability. Moreover, with the established ML models as a readout for the chemical screen, we identify a CDK8 inhibitor that can further improve the cell resistance to the overdose of CHIR. Together, this study indicates that artificial intelligence is able to guide and iteratively optimize PSC differentiation to achieve consistently high efficiency across cell lines and batches, providing a better understanding and rational modulation of the differentiation process for functional cell manufacturing in biomedical applications.
Yang, X., Chen, D., Sun, Q., Wang, Y., Xia, Y., Yang, J., Lin, C., Dang, X., Cen, Z., Liang, D., Wei, R., Xu, Z., Xi, G., Xue, G., Ye, C., Wang, L.-P., Zou, P., Wang, S., Rivera-Fuentes, P., Püntener, S., Chen, Z., Liu, Y.*, Zhang, J.* and Zhao, Y.* (2023) Cell Discov. 9, 53.
Subcellular protein-protein interactions (PPIs) are essential to understanding the mechanism of diverse cellular signaling events and the pathogenesis of diseases. Herein, we report an integrated APEX proximity labeling and chemical cross-linking coupled with mass spectrometry (CXMS) platform named APEX-CXMS for spatially resolved subcellular interactome profiling in a high-throughput manner. APEX proximity labeling rapidly captures subcellular proteomes, and the highly reactive chemical cross-linkers can capture weak and dynamic interactions globally without extra genetic manipulation. APEX-CXMS was first applied to mitochondria and identified 653 pairs of interprotein cross-links. Six pairs of new interactions were selected and verified by coimmunoprecipitation, the mammalian two-hybrid system, and surface plasmon resonance method. Besides, our approach was further applied to the nucleus, capturing 336 pairs of interprotein cross-links with approximately 94% nuclear specificity. APEX-CXMS thus provides a simple, fast, and general alternative to map diverse subcellular PPIs.
Sun, M., Yuan, F., Tang, Y., Zou, P.* and Lei, X.* (2022) Anal. Chem. 94, 14878-14888.
Error-free mitosis depends on accurate chromosome attachment to spindle microtubules via a fine structure called the centromere that is epigenetically specified by the enrichment of CENP-A nucleosomes. Centromere maintenance during mitosis requires CENP-A-mediated deposition of constitutive centromere-associated network that establishes the inner kinetochore and connects centromeric chromatin to spindle microtubules during mitosis. Although previously proposed to be an adaptor of retinoic acid receptor, here, we show that CENP-R synergizes with CENP-OPQU to regulate kinetochore-microtubule attachment stability and ensure accurate chromosome segregation in mitosis. We found that a phospho-mimicking mutation of CENP-R weakened its localization to the kinetochore, suggesting that phosphorylation may regulate its localization. Perturbation of CENP-R phosphorylation is shown to prevent proper kinetochore-microtubule attachment at metaphase. Mechanistically, CENP-R phosphorylation disrupts its binding with CENP-U. Thus, we speculate that Aurora B-mediated CENP-R phosphorylation promotes the correction of improper kinetochore-microtubule attachment in mitosis. As CENP-R is absent from yeast, we reasoned that metazoan evolved an elaborate chromosome stability control machinery to ensure faithful chromosome segregation in mitosis.
Sedzro, D. M., Yuan, X., Mullen, M., Ejaz, U., Yang, T., Liu, X., Song, X., Tang, Y.-C., Pan, W., Zou, P., Gao, X., Wang, D., Wang, Z., Dou, Z.*, Liu, X.* and Yao, X.* (2022) J. Mol. Cell Biol. 14, mjac051.
The identification of the structure of protein complexes in the subcellular niche of cells is necessary to understand their diverse functions. In this study, we developed a suborganelle proteome labeling assisted in vivo cross-linking (SubPiXL) strategy to identify regional protein conformations and interactions in living cells. Due to the mitochondria’s functional importance and well-defined compartmental partitions, the specific conformations and interactome of protein complexes located in the mitochondrial matrix were identified. Compared to the commonly used approach of organelle isolation followed by intact mitochondria cross-linking, our method achieved a more refined spatial characterization for the subcompartment of the cellular organelle. Additionally, this approach avoided cross-contamination and cell microenvironment disruption during organelle isolation. As such, we achieved 73% selectivity for mitochondria and 98% specificity of known suborganelle annotation for the mitochondrial matrix and accessible inner membrane. Meanwhile, more protein–protein interactions (PPIs) with high dynamics were captured, resulting in a 1.67-fold increase in the number of PPI identifications in 1/11th of the time. On the basis of these structural cross-links and the specific characterization of the interactome and conformation, the structural dynamics targeted in the mitochondrial matrix were delineated. Mitochondrial matrix-restricted information for proteins with multisubcellular localizations was then clarified. In summary, SubPiXL is a promising technique for the investigation of suborganelle-resolved protein conformation and interaction analysis and contributes to a better understanding of structure-derived functions.
An, Y., Zhao, Q.*, Gong, Z., Zhao, L., Li, Y., Liang, Z., Zou, P.*, Zhang, Y. and Zhang, L.* (2022) Anal. Chem. 94, 12051-12059.
The back-propagating action potential (bpAP) is crucial for neuronal signal integration and synaptic plasticity in dendritic trees. Its properties (velocity and amplitude) can be affected by dendritic morphology. Due to limited spatial resolution, it has been difficult to explore the specific propagation process of bpAPs along dendrites and examine the influence of dendritic morphology, such as the dendrite diameter and branching pattern, using patch-clamp recording. By taking advantage of Optopatch, an all-optical electrophysiological method, we made detailed recordings of the real-time propagation of bpAPs in dendritic trees. We found that the velocity of bpAPs was not uniform in a single dendrite, and the bpAP velocity differed among distinct dendrites of the same neuron. The velocity of a bpAP was positively correlated with the diameter of the dendrite on which it propagated. In addition, when bpAPs passed through a dendritic branch point, their velocity decreased significantly. Similar to velocity, the amplitude of bpAPs was also positively correlated with dendritic diameter, and the attenuation patterns of bpAPs differed among different dendrites. Simulation results from neuron models with different dendritic morphology corresponded well with the experimental results. These findings indicate that the dendritic diameter and branching pattern significantly influence the properties of bpAPs. The diversity among the bpAPs recorded in different neurons was mainly due to differences in dendritic morphology. These results may inspire the construction of neuronal models to predict the propagation of bpAPs in dendrites with enormous variation in morphology, to further illuminate the role of bpAPs in neuronal communication.
Tian, W.#, Peng, L.#, Zhao, M.#, Tao, L.*, Zou, P.* and Zhang, Y.* (2022) Neurosci. Bull. in press, DOI: 10.1007/s12264-022-00931-9
Action potentials (APs) in neurons are generated at the axon initial segment (AIS). AP dynamics, including initiation and propagation, are intimately associated with neuronal excitability and neurotransmitter release kinetics. Most learning and memory studies at the single-neuron level have relied on the use of animal models, most notably rodents. Here, we studied AP initiation and propagation in cultured hippocampal neurons from Sprague-Dawley (SD) rats and C57BL/6 (C57) mice with genetically encoded voltage indicator (GEVI)-based voltage imaging. Our data showed that APs traveled bidirectionally in neurons from both species; forward-propagating APs (fpAPs) had a different speed than backpropagating APs (bpAPs). Additionally, we observed distinct AP propagation characteristics in AISs emerging from the somatic envelope compared to those originating from dendrites. Compared with rat neurons, mouse neurons exhibited higher bpAP speed and lower fpAP speed, more distally located ankyrin G (AnkG) in AISs, and longer Nav1.2 lengths in AISs. Moreover, during AIS plasticity, AnkG and Nav1.2 showed distal shifts in location and shorter lengths of labeled AISs in rat neurons; in mouse neurons, however, they showed a longer AnkG-labeled length and more distal Nav1.2 location. Our findings suggest that hippocampal neurons in SD rats and C57 mice may have different AP propagation speeds, different AnkG and Nav1.2 patterns in the AIS, and different AIS plasticity properties, indicating that comparisons between these species must be carefully considered.
Chen, Z., Peng, L., Zhao, M., Tao, L.*, Zou, P.* and Zhang, Y.* (2022) Zool. Res. 43, 615-633.
Recent work has made it clear that pericentriolar material (PCM), the matrix of proteins surrounding centrioles, contributes to most functions of centrosomes. Given the occurrence of centrosome amplification in most solid tumors and the unconventional survival of these tumor cells, it is tempting to hypothesize that gel-like mitotic PCM would cluster extra centrosomes to defend against mitotic errors and increase tumor cell survival. However, because PCM lacks an encompassing membrane, is highly dynamic, and is physically connected to centrioles, few methods can decode the components of this microscale matrix. In this study, we took advantage of differential labeling between two sets of APEX2-centrosome reactions to design a strategy for acquiring the PCM proteome in living undisturbed cells without synchronization treatment, which identified 392 PCM proteins. Localization of ubiquitination promotion proteins away from PCM was a predominant mechanism to maintain the large size of PCM for centrosome clustering during mitosis in cancer cells. Depletion of PCM gene kinesin family member 20A (KIF20A) caused centrosome clustering failure and apoptosis in cancer cells in vitro and in vivo. Thus, our study suggests a strategy for targeting a wide range of tumors exhibiting centrosome amplification and provides a proteomic resource for future mining of PCM proteins.
Xie, B.#, Pu, Y.#, Yang, F., Chen, W., Yue, W., Ma, J., Zhang, N., Jiang, Y., Wu, J., Lin, Y., Liang, X., Wang, W., Zou, P.* and Li, M.* (2022) Cancer Res. 82, 2576-2592.
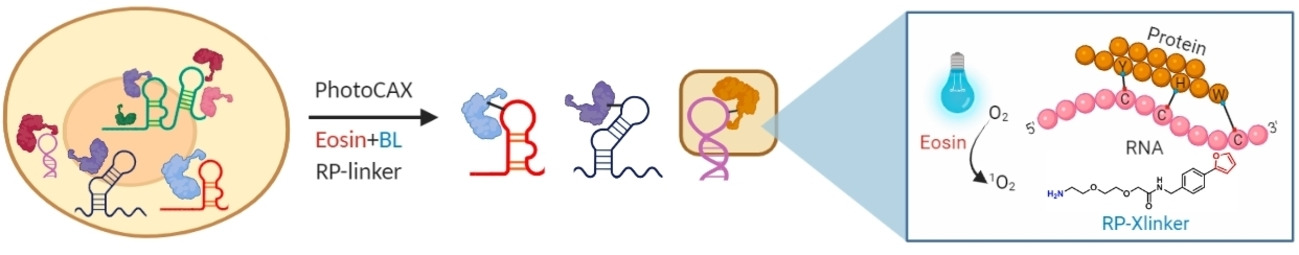
The dynamic interactions between RNAs and proteins play crucial roles in regulating diverse cellular processes. Proteome-wide characterization of these interactions in their native cellular context remains desirable but challenging. Herein, we developed a photocatalytic crosslinking (PhotoCAX) strategy coupled with mass spectrometry (PhotoCAX-MS) and RNA sequencing (PhotoCAX-seq) for the study of the composition and dynamics of protein-RNA interactions. By integrating the blue light-triggered photocatalyst with a dual-functional RNA–protein crosslinker (RP-linker) and the phase separation-based enrichment strategy, PhotoCAX-MS revealed a total of 2044 RBPs in human HEK293 cells. We further employed PhotoCAX to investigate the dynamic change of RBPome in macrophage cells upon LPS-stimulation, as well as the identification of RBPs interacting directly with the 5′ untranslated regions of SARS-CoV-2 RNA.
Luo, H.#, Tang, W.#, Liu, H., Zeng, X., Ngai, W. S. C., Gao, R., Li, H., Li, R., Zheng, H., Guo, J., Qin, F., Wang, G., Li, K., Fan, X.*, Zou, P.* and Chen, P. R.* (2022) Angew. Chem. Int. Ed. Engl. 61, e202202008.
Liquid–liquid phase separation (LLPS) of SynGAP and PSD-95, two abundant proteins that interact in the postsynaptic density (PSD) of neurons, has been implicated in modulating SynGAP PSD enrichment in excitatory synapses. However, the underlying regulatory mechanisms remain enigmatic. Here we report that O-GlcNAcylation of SynGAP acts as a suppressor of LLPS of the SynGAP/PSD-95 complex. We identified multiple O-GlcNAc modification sites for the endogenous SynGAP isolated from rat brain and the recombinantly expressed protein. Protein semisynthesis was used to generate site-specifically O-GlcNAcylated forms of SynGAP, and in vitro and cell-based LLPS assays demonstrated that T1306 O-GlcNAc of SynGAP blocks the interaction with PSD-95, thus inhibiting LLPS. Furthermore, O-GlcNAcylation suppresses SynGAP/PSD-95 LLPS in a dominant-negative manner, enabling sub-stoichiometric O-GlcNAcylation to exert effective regulation. We also showed that O-GlcNAc-dependent LLPS is reversibly regulated by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). These findings demonstrate that OGT- and OGA-catalysed O-GlcNAc cycling may serve as an LLPS-regulating post-translational modification.
Lv, P.#, Du, Y.#, He, C.#, Peng, L., Zhou, X., Wan, Y., Zeng, M., Zhou, W., Zou, P., Li, C., Zhang, M., Dong, S.* and Chen, X.* (2022) Nat. Chem. 14, 831.
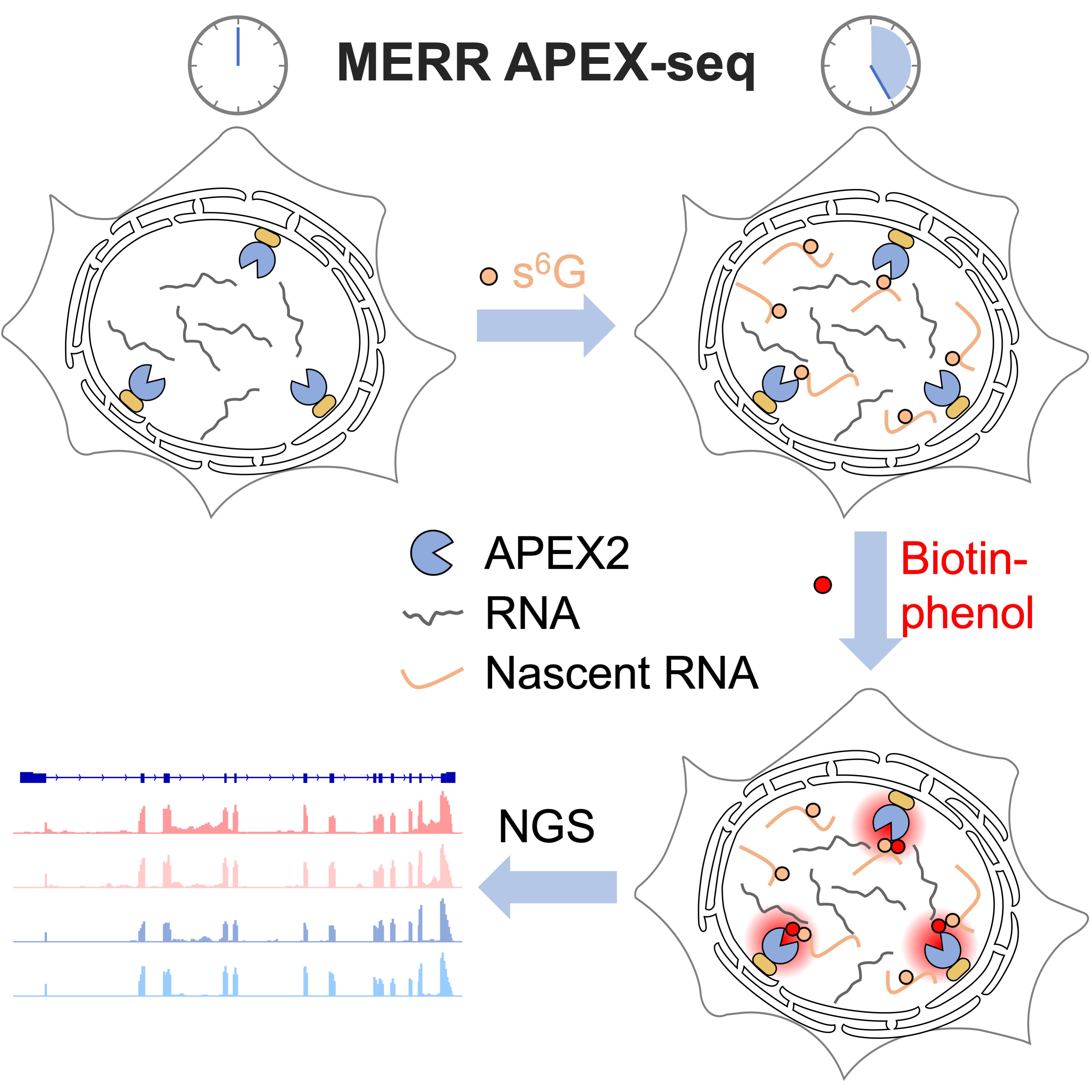
The spatial arrangement of newly synthesized transcriptome in eukaryotic cells underlies various biological processes including cell proliferation and differentiation. In this study, we combine metabolic incorporation of electron-rich ribonucleosides (e.g. 6-thioguanosine and 4-thiouridine) with peroxidase-mediated proximity-dependent RNA labeling technique (APEX-seq) to develop a sensitive method, termed MERR APEX-seq, for selectively profiling newly transcribed RNAs at specific subcellular locations in live cells. We demonstrate that MERR APEX-seq is 20-fold more efficient than APEX-seq and offers both high spatial specificity and high coverage in mitochondrial matrix. At endoplasmic reticulum membrane, 91% of the transcripts captured by MERR APEX-seq encode for secretory pathway proteins, thus demonstrating the high spatial specificity of MERR APEX-seq in open subcellular compartment. Application of MERR APEX-seq to the nuclear lamina of human cells reveals a local transcriptome of 1012 RNAs, many of which encode for nuclear proteins involved in histone modification, chromosomal structure maintenance and RNA processing.
Li, R., Zou, Z., Wang, W. and Zou, P.* (2022) Cell Chem. Biol. 29, 1218.
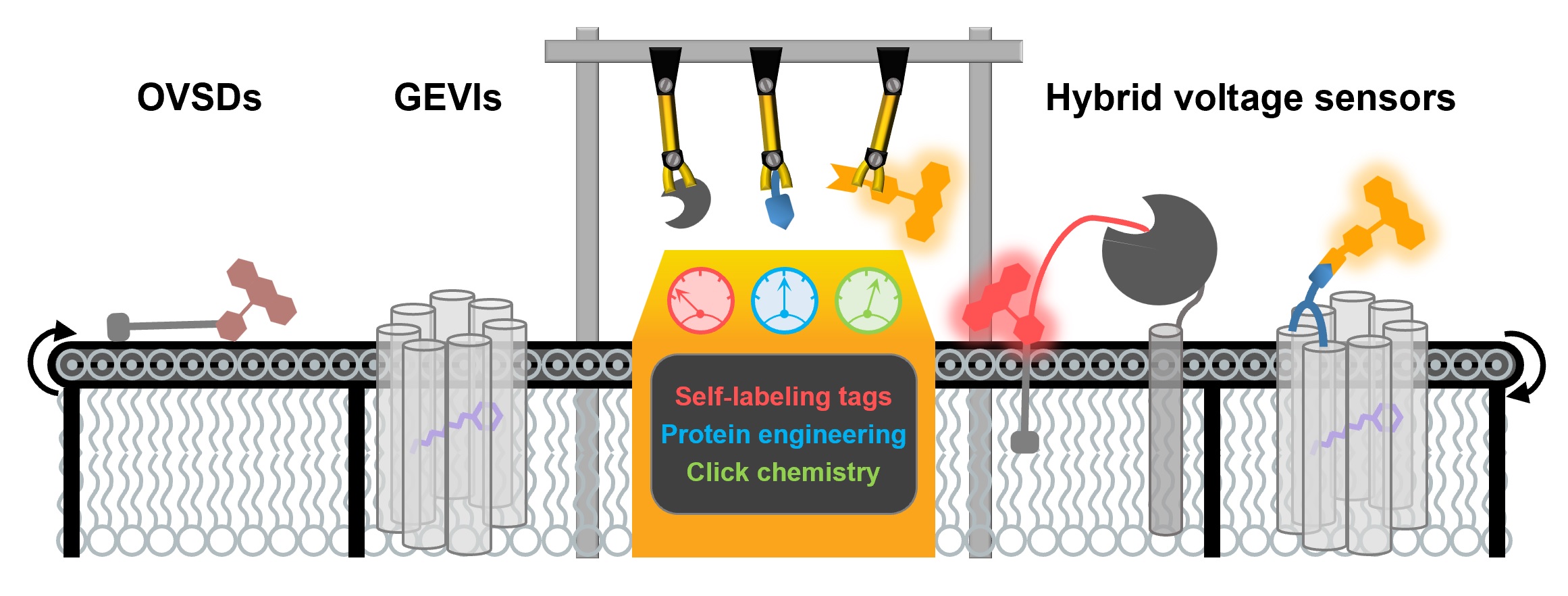
Membrane potential is an indispensable biophysical signal in neurobiology. Imaging neuronal electrical signals with fluorescent indicators allows for non-invasive recording at high spatial resolution. Over the past decades, both genetically encoded voltage indicators (GEVIs) and organic voltage sensing dyes (OVSDs) have been developed to achieve imaging membrane potential dynamics in cultured neurons and in vivo. More recently, hybrid voltage indicators have gained increasing attention due to their superior fluorescent quantum yield and photostability as compared to conventional GEVIs. In this mini-review, we summarize the design, characterization and biological applications of hybrid voltage indicators, and discuss future improvements.
Liu, S.#, Yang, J.# and Zou, P.* (2021) J. Neurosci. Methods 363, 109348.
Mammalian cell nuclei contain copper, and cancer cells are known to accumulate aberrantly high copper levels, yet the mechanisms underlying nuclear accumulation and copper's broader functional significance remain poorly understood. Here, by combining APEX2-based proximity labeling focused on the copper chaperone Atox1 with mass spectrometry we identified a previously unrecognized nuclear copper binding protein, Cysteine-rich protein 2 (CRIP2), that interacts with Atox1 in the nucleus. We show that Atox1 transfers copper to CRIP2, which induces a change in CRIP2's secondary structure that ultimately promotes its ubiquitin-mediated proteasomal degradation. Finally, we demonstrate that depletion of CRIP2-as well as copper-induced CRIP2 degradation-elevates ROS levels and activates autophagy in H1299 cells. Thus, our study establishes that CRIP2 as an autophagic suppressor protein and implicates CRIP2-mediated copper metabolism in the activation of autophagy in cancer cells.
Chen, L., Li, N., Zhang, M., Sun, M., Bian, J., Yang, B., Li, Z., Wang, J., Li, F., Shi, X., Wang, Y., Yuan, F., Zou, P., Shan, C. and Wang, J.* (2021) Angew. Chem. Int. Ed. Engl. 60, 25346-25355.
As a common feature of tumors, chromosomal instability (CIN) not only forces carcinomatous evolution, but also loads cancer cells with extra pressure through a robust imbalance of genome patterning that may be used for cancer treatment. Errors in cytokinesis increase CIN, so cytokinesis components are valuable targets for treating cancer. However, due to the short time span and confined space of cytokinesis bridges, profiling cytokinesis fac- tors is challenging. Taking advantage of engineered ascorbate peroxidase (APEX2), we established a cytokinesis bridge-APEX reaction in living cells. A total of 218 cytokinesis bridge proteins were identified with high relia- bility. Knockdown of cytokinesis bridge genes generated micronuclei that activate the cGAS-pathway and cause apoptosis in cancer cells bearing high CIN rather than low CIN. Thus, our study proposes a strategy for killing high-CIN tumors regardless of tumor type, and provides a proteome resource of cytokinetic bridges for future research.
Xie, B., Liang, X., Yue, W., Ma, J., Li, X., Zhang, N., Wang, P., Liu, C., Shi, X., Qiao, J., Zou, P. and Li, M.* (2021). Fundamental Research 1, 752-766.
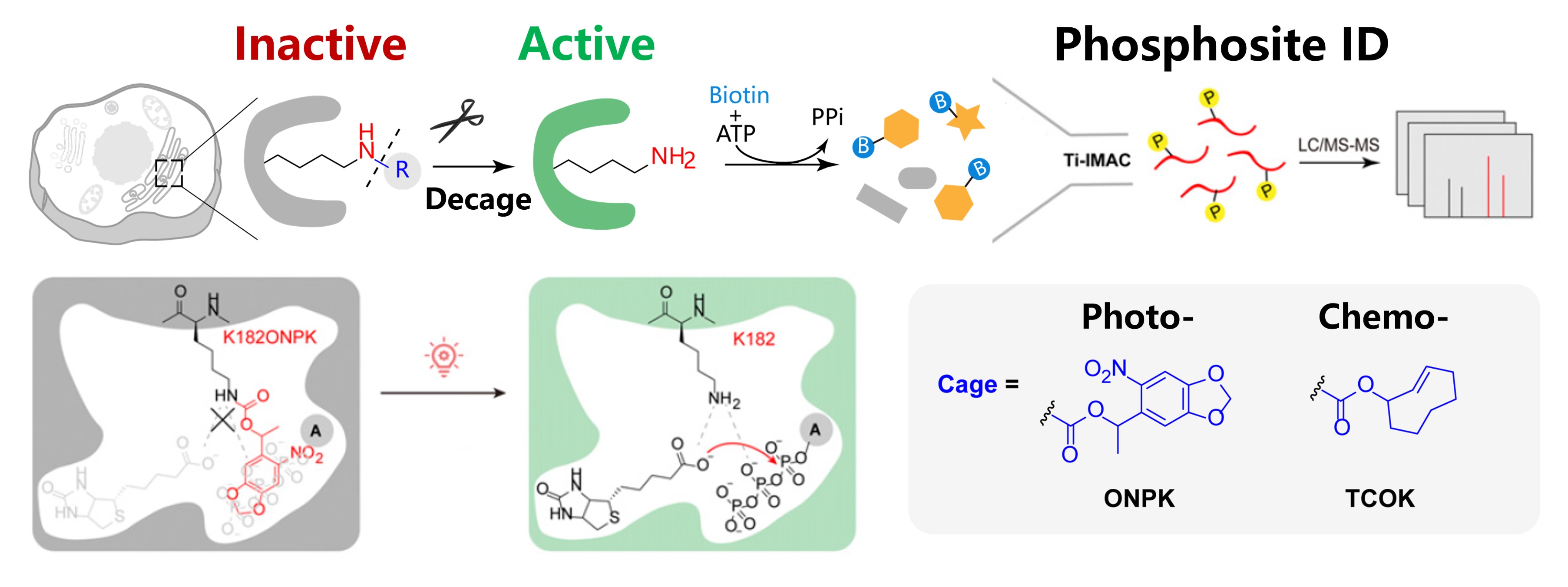
Proteome-wide profiling of protein phosphorylation has been widely used to reveal the underlying mechanism of diverse cellular signaling events. Yet, characterizing subcellular phosphoproteome with high spatial–temporal resolution has remained challenging. Herein, we developed a subcellular-specific uncaging-assisted biotinylation and mapping of phosphoproteome (SubMAPP) strategy to monitor the phosphorylation dynamics of subcellular proteome in living cells and animals. Our method capitalizes on the genetically encoded bioorthogonal decaging strategy, which enables the rapid activation of subcellular localized proximity labeling biotin ligase through either light illumination or small-molecule triggers. By further adopting an integrated orthogonal pull-down strategy with quantitative mass spectrometry, SubMAPP allowed for the investigation of subcellular phosphoproteome dynamics, revealing the altered phosphorylation patterns of endoplasmic reticulum (ER) luminal proteins under ER stress. Finally, we further expanded the scope of the SubMAPP strategy to primary neuron culture and living mice.
Liu, Y.#, Zeng, R.#, Wang, R., Weng, Y., Wang, R., Zou, P.* and Chen, P. R.* (2021). Proc. Natl. Acad. Sci. U. S. A. 118, e2025299118.
Delayed neurocognitive recovery (dNCR) is a major complication after anesthesia and surgery in older adults. Alpha-synuclein (α-syn; encoded by the gene, SNCA) has recently been shown to play an important role in hippocampus-dependent working memory. Aggregated forms of α-syn are associated with multiple neurotoxic mechanisms, such as mitochondrial dysfunction and cell death. In this study, we found that blocking α-syn improved both mitochondrial function and mitochondria-dependent neuronal apoptosis in a mouse model of dNCR. Various forms of α-syn (including total α-syn, phosphorylated-Ser129-α-syn, and oligomers) were upregulated in hippocampal tissue and extracted mitochondria after surgical challenge. Clenbuterol is a novel transcription modulator of Scna. Clenbuterol significantly attenuated surgery-induced progressive accumulation of various toxic α-syn forms in the hippocampus, as well as mitochondrial damage and memory deficits in aged mice following surgery. We also observed excessive mitochondrial α-syn accumulation and increased mitochondria-mediated apoptosis in vitro using nerve growth factor-differentiated PC12 cells and primary hippocampal neurons exposed to lipopolysaccharide. To further validate the neuroprotective effect of α-syn inhibition, we used a lentiviral Snca-shRNA (Lv-shSnca) to knockdown Snca. Of note, Lv-shSnca transfection significantly inhibited neuronal apoptosis mediated by the mitochondrial apoptosis pathway in neurons exposed to lipopolysaccharide. This α-syn inhibition improved the disruption to mitochondrial morphology and function, as well as decreased levels of apoptosis. Our results suggest that targeting pathological α-syn may achieve neuroprotection through regulation of mitochondrial homeostasis and suppression of apoptosis in the aged hippocampus, further strengthening the therapeutic potential of targeting α-syn for dNCR.
Li, Y., Yuan, Y., Li, Y., Han, D., Liu, T., Yang, N., Mi, X., Hong, J., Liu, K., Song, Y., He, J., Zhou, Y., Han, Y., Shi, C., Yu, S., Zou, P., Guo, X.* and Li, Z.* (2021). Oxid. Med. Cell Longev. 2021, 5572899.
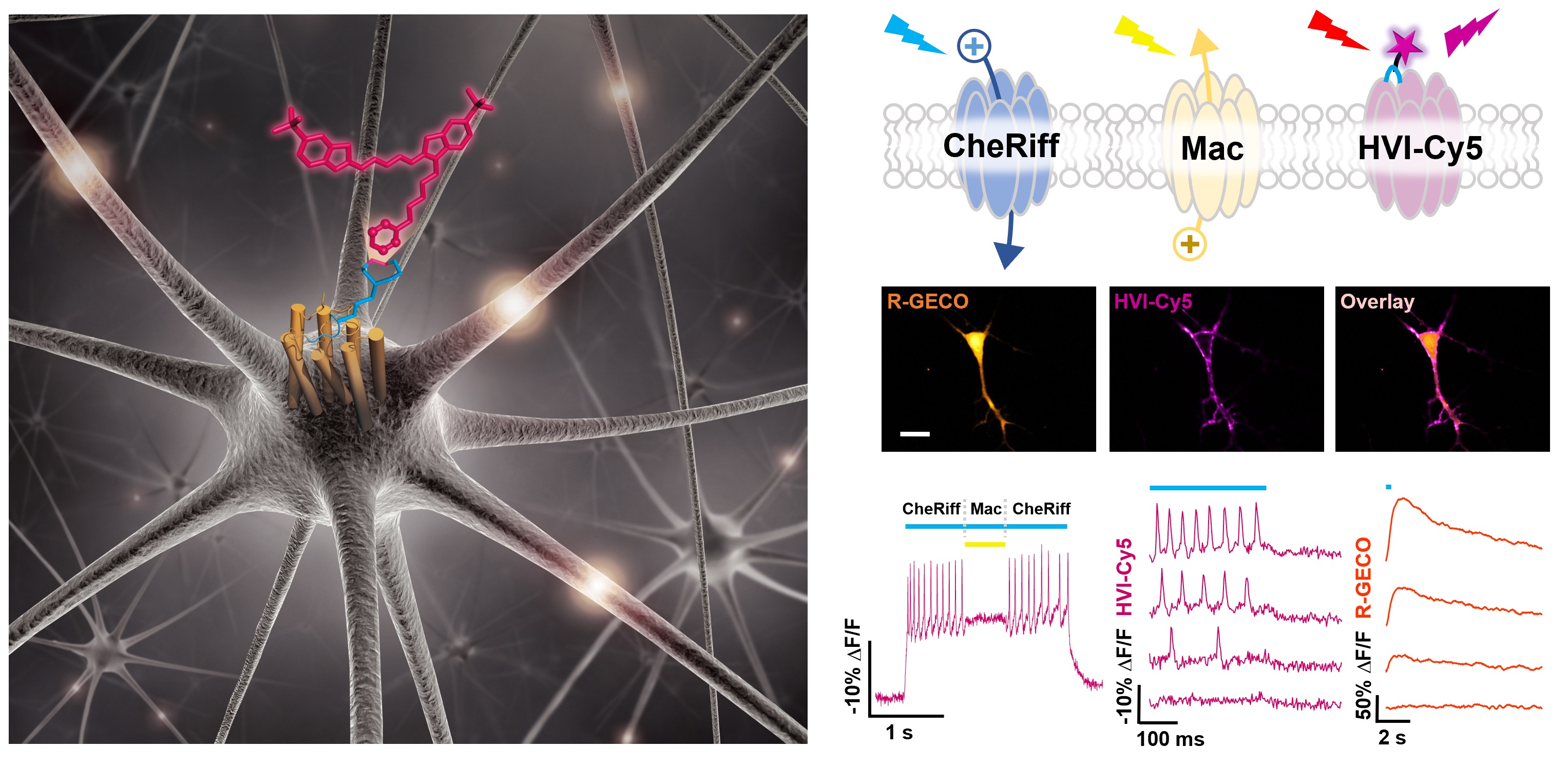
Membrane potential is a key aspect of cellular signalling and is dynamically regulated by an array of ion-selective pumps and channels. Fluorescent voltage indicators enable non-invasive optical recording of the cellular membrane potential with high spatial resolution. Here, we report a palette of bright and sensitive hybrid voltage indicators (HVIs) with fluorescence intensities sensitive to changes in membrane potential via electrochromic Förster resonance energy transfer. Enzyme-mediated site-specific incorporation of a probe, followed by an inverse-electron-demand Diels–Alder cycloaddition, was used to create enhanced voltage-sensing rhodopsins with hybrid dye–protein architectures. The most sensitive indicator, HVI-Cy3, displays high voltage sensitivity (−39% ΔF/F0 per 100 mV) and millisecond response kinetics, enabling optical recording of action potentials at a sampling rate of 400 Hz over 10 min across a large neuronal population. The far-red indicator HVI-Cy5 could be paired with optogenetic actuators and green/red-emitting fluorescent indicators, allowing an all-optical investigation of neuronal electrophysiology.
Liu, S.#, Lin, C.#, Xu, Y.#, Luo, H., Peng, L., Zeng, X., Zheng, H., Chen, P. R.* and Zou, P.* (2021). Nat. Chem. 13, 472-479.
Critical early steps in human embryonic development include polarization of the inner cell mass, followed by formation of an expanded lumen that will become the epiblast cavity. Recently described three-dimensional (3D) human pluripotent stem cell-derived cyst (hPSC-cyst) structures can replicate these processes. To gain mechanistic insights into the poorly understood machinery involved in epiblast cavity formation, we interrogated the proteomes of apical and basolateral membrane territories in 3D human hPSC-cysts. APEX2-based proximity bioinylation, followed by quantitative mass spectrometry, revealed a variety of proteins without previous annotation to specific membrane subdomains. Functional experiments validated the requirement for several apically enriched proteins in cyst morphogenesis. In particular, we found a key role for the AP-1 clathrin adaptor complex in expanding the apical membrane domains during lumen establishment. These findings highlight the robust power of this proximity labeling approach for discovering novel regulators of epithelial morphogenesis in 3D stem cell-based models.
Wang, S., Lin, C.-W., Carleton, A. E., Cortez, C. L., Johnson, C., Taniguchi, L. E., Sekulovski, N., Townshend, R. F., Basrur, V., Nesvizhskii, A. I., Zou, P., Fu, J.*, Gumucio, D. L.*, Duncan, M. C.* and Taniguchi, K.* (2021). Sci. Adv. 7, eabd8407.
Cells mitigate ER stress through the unfolded protein response (UPR). Here, we report formation of ER whorls as an effector mechanism of the ER stress response. We found that strong ER stress induces formation of ER whorls, which contain ER-resident proteins such as the Sec61 complex and PKR-like ER kinase (PERK). ER whorl formation is dependent on PERK kinase activity and is mediated by COPII machinery, which facilitates ER membrane budding to form tubular-vesicular ER whorl precursors. ER whorl precursors then go through Sec22b-mediated fusion to form ER whorls. We further show that ER whorls contribute to ER stress-induced translational inhibition by possibly modulating PERK activity and by sequestering translocons in a ribosome-free environment. We propose that formation of ER whorls reflects a new type of ER stress response that controls inhibition of protein translation.
Ke, M., Yuan, X., He, A., Yu, P., Chen, W., Shi, Y., Hunter, T., Zou, P. and Tian, R.* (2021). Nat. Commun. 12, 71.
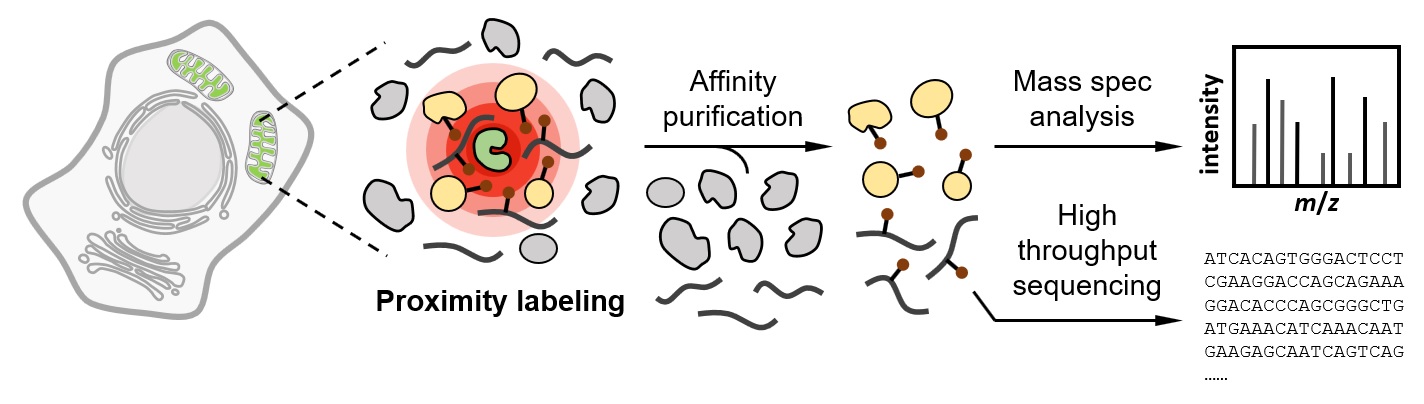
The subcellular organization of proteins and RNA molecules is crucial for their proper functions. Over the past decade, both ligase-mediated and peroxidase-mediated proximity labeling (PL) techniques have been developed to map biomolecules at near-nanometer spatial resolution and subminute temporal resolution. These methods are shedding light on the spatial arrangement of proteome and transcriptome in their native context. Here, we review the recent evolution and applications of PL techniques, compare and contrast the two classes of methods, and highlight emerging trends and future opportunities.
Zhou, Y. and Zou, P.* (2021). Curr. Opin. Chem. Biol. 60, 30-38.
Cells mitigate ER stress through the unfolded protein response (UPR). Here, we report formation of ER whorls as an effector mechanism of the ER stress response. We found that strong ER stress induces formation of ER whorls, which contain ER-resident proteins such as the Sec61 complex and PKR-like ER kinase (PERK). ER whorl formation is dependent on PERK kinase activity and is mediated by COPII machinery, which facilitates ER membrane budding to form tubular-vesicular ER whorl precursors. ER whorl precursors then go through Sec22b-mediated fusion to form ER whorls. We further show that ER whorls contribute to ER stress-induced translational inhibition by possibly modulating PERK activity and by sequestering translocons in a ribosome-free environment. We propose that formation of ER whorls reflects a new type of ER stress response that controls inhibition of protein translation.
Xu, F., Du, W., Zou, Q., Wang, Y., Zhang, X., Xing, X., Li, Y., Zhang, D., Wang, H., Zhang, W., Hu, X., Liu, X., Liu, X., Zhang, S., Yu, J., Fang, J., Li, F., Zhou, Y., Yue, T., Mi, N., Deng, H., Zou, P., Chen, X., Yang, X.* and Yu, L.* (2021). Cell Res. 31, 141-156.
2016 - 2020
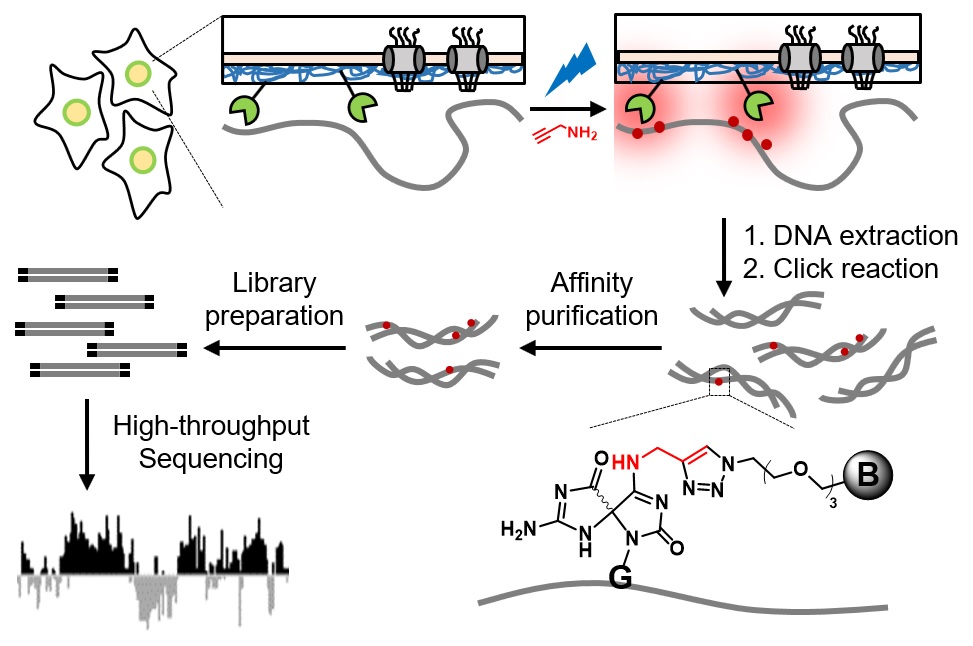
The spatial arrangement of chromosome within the nucleus is intimately linked to genome function and gene expression regulation. Existing genome‐wide mapping methods to study chromosome organization often rely on chemically crosslinking DNA with protein baits, which raises concerns of introducing artefacts during cellular fixation. Herein, we developed a novel proximity‐dependent DNA labeling method based on the chromophore‐assisted nucleobase photooxidation. By genetically targeting a photosensitizer protein to specific subnuclear locations, we achieved blue light‐activated labeling of local DNA with a bioorthogonal functional handle, which allowed subsequent affinity purification and sequence identification via next‐generation sequencing. When applied to the nuclear lamina in human embryonic kidney 293T cells, our method revealed lamina‐associated domains (LADs) that cover 37.6% of the genome. These LADs overlap with heterochromatin hallmarks including histone 3 lysine 9 dimethylation (H3K9me2) and are depleted with CpG islands. This simple labeling method avoids the harsh treatment of chemical crosslinking and is generally applicable for the genome‐wide high‐resolution mapping of the spatial chromosome organization in living cells.
Ding, T.#, Zhu, L.#, Fang, Y., Liu, Y., Tang, W. and Zou, P.* (2020). Angew. Chem. Int. Ed. Engl. 59, 22933-22937.
Alk-Ph is a clickable APEX2 substrate developed for spatially restricted protein/RNA labeling in intact yeast cells. Alk-Ph is more water soluble and cell wall permeable than biotin-phenol substrate, allowing more efficient profiling of the subcellular proteome in microorganisms. We describe the protocol for Alk-Ph probe synthesis, APEX2 expression, and protein/RNA labeling in yeast and the workflow for quantitative proteomic experiments and data analysis. Using the yeast mitochondria as an example, we provide guidelines to achieve high-resolution mapping of subcellular yeast proteome and transcriptome. For complete details on the use and execution of this protocol, please refer to Li et al. (2020).
Li, Y., Liu, K., Zhou, Y., Yang, J.* and Zou, P.* (2020). STAR Protoc. 1, 100137.
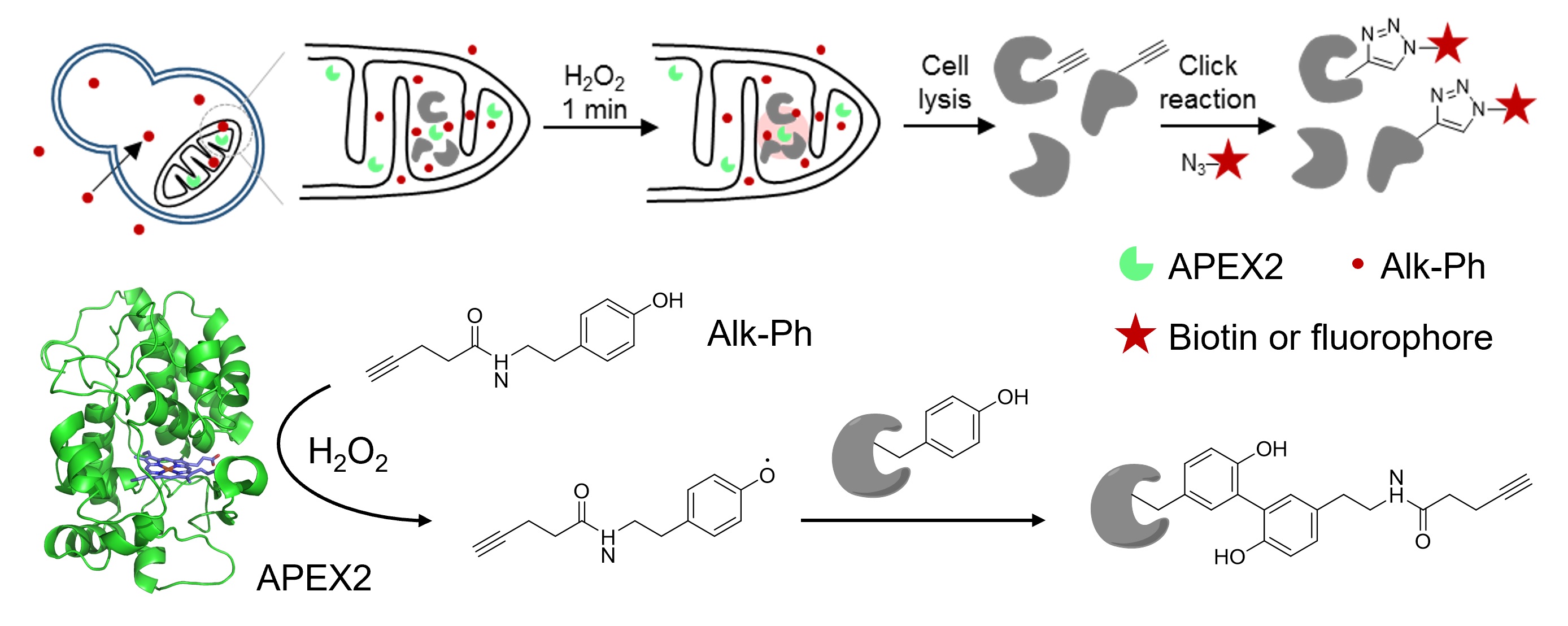
The engineered ascorbate peroxidase (APEX) is a powerful tool for the proximity-dependent labeling of proteins and RNAs in live cells. Although widely employed in mammalian cells, APEX applications in microorganisms have been hampered by the poor labeling efficiency of its biotin-phenol (BP) substrate. In this study, we sought to address this challenge by designing and screening a panel of alkyne-functionalized substrates. Our best probe, Alk-Ph, substantially improves APEX labeling efficiency in intact yeast cells, as it is more cell wall-permeant than BP. Through a combination of protein-centric and peptide-centric chemoproteomic experiments, we have identified 165 proteins with a specificity of 94% in the yeast mitochondrial matrix. In addition, we have demonstrated that Alk-Ph is useful for proximity-dependent RNA labeling in yeast, thus expanding the scope of APEX-seq. We envision that this improved APEX labeling strategy would set the stage for the large-scale mapping of spatial proteome and transcriptome in yeast.
Li, Y., Tian, C., Liu, K., Zhou, Y., Yang, J.* and Zou, P.* (2020). Cell Chem. Biol. 27, 858-865.
Little is known about the underlying mechanisms of the similarities in the core features of postoperative delirium (POD) and α-synuclein (α-syn)-related cognitive disorders. We herein investigated associations between fluctuated levels of exosomal α-syn in the plasma and POD presentation in geriatric hip fracture patients. Exosome α-syn release in plasma may be associated with the POD development which might be due to systemic inflammation.
Yuan, Y.#, Li, Z.#, Yang, N.#, Han, Y., Ji, X., Han, D., Wang, X., Li, Y., Liu, T., Yuan, F., He, J., Liu, Y., Ni, C., Zou, P., Wang, G.*, Guo, X.* and Zhou, Y.* (2020). Front Aging Neurosci. 12, 67.
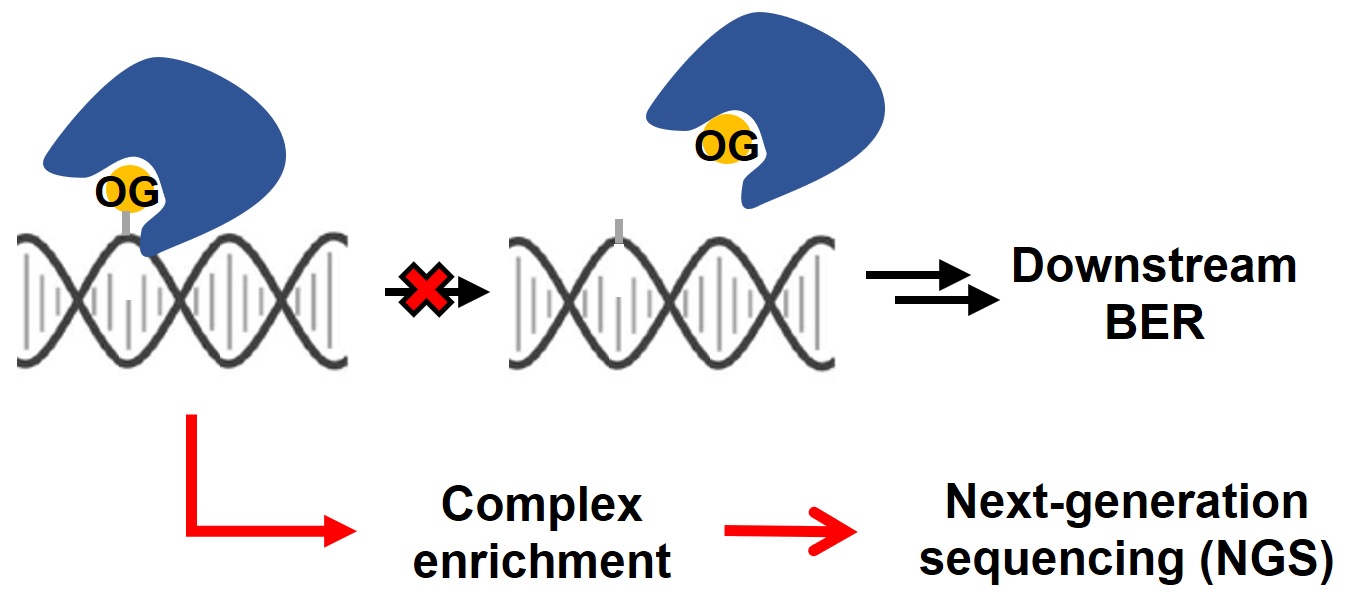
The occurrence of 8-oxo-7,8-dihydroguanine (OG) in the genome, as one of the major DNA oxidative damages, has been implicated in an array of biological processes, ranging from mutagenesis to transcriptional regulation. Genome-wide mapping of oxidative damages could shed light on the underlying cellular mechanism. In the present study, we engineered the hOGG1 enzyme, a primary 8-oxoguanine DNA glycosylase, into a guanine oxidation-profiling tool. Our method, called enTRAP-seq, successfully identified more than 1,400 guanine oxidation sites in the mouse embryonic fibroblast genome. These OG peaks were enriched in open chromatin regions and regulatory elements including pro-moters, 5’ untranslated regions and CpG islands. Collectively, we present a simple and generalizable approach for the genome-wide profiling of DNA damages with high sensitivity and specificity.
Fang, Y. and Zou, P.* (2020). Biochemistry 59, 85-89.
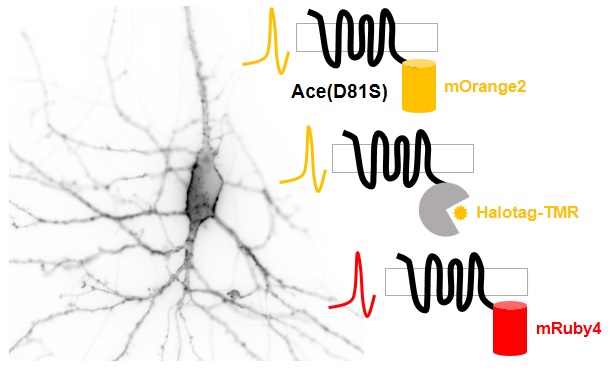
Genetically encoded voltage indicators (GEVIs) allow optical recording of neuronal activities with high spatial resolution. While most existing GEVIs emit in the green range, red-shifted GEVIs are highly sought after because they would enable simultaneous stimulation and recording of neuronal activities when paired with optogenetic actuators, or two-color imaging of signaling and neuronal activities when used along with GFP-based indicators. In this study, we present several improved red-shifted GEVIs based on the electrochromic Förster resonance energy transfer (eFRET) between orange/red fluorescent proteins/dyes and rhodopsin mutants. Through structure-guided mutagenesis and cell-based sensitivity screening, we identified a mutant rhodopsin with a single mutation that exhibited more than 2-fold improvement in voltage sensitivity. Notably, this mutation has been independently discovered by Pieribone et al. (Pieribone, V. A. et al. Nat Methods 2018, 15 (12), 1108−1116). In cultured rat hippocampal neurons, our sensors faithfully reported action potential waveforms and subthreshold activities. We also demonstrated that this mutation could enhance the sensitivity of hybrid indicators, thus providing insights for future development.
Xu, Y.#, Deng, M.#, Zhang, S.#, Yang, J.#, Peng, L., Chu, J.* and Zou, P.* (2019). ACS Chem. Neurosci. 10, 4768-4775.
Biological macromolecules (proteins, nucleic acids, polysaccharides, etc.) are the building blocks of life, which constantly undergo chemical modifications that are often reversible and spatial-temporally regulated. These dynamic properties of chemical modifications play fundamental roles in physiological processes as well as pathological changes of living systems. The Major Research Project (MRP) funded by the National Natural Science Foundation of China (NSFC)-"Dynamic modifications of biomacromolecules: mechanism and chemical interventions" aims to integrate cross-disciplinary approaches at the interface of chemistry, life sciences, medicine, mathematics, material science and information science with the following goals: (i) developing specific labeling techniques and detection methods for dynamic chemical modifications of biomacromolecules, (ii) analyzing the molecular mechanisms and functional relationships of dynamic chemical modifications of biomacromolecules, and (iii) exploring biomacromolecules and small molecule probes as potential drug targets and lead compounds.
Wang, C., Zou, P., Yang, C., Liu, L., Cheng, L., He, X., Zhang, L., Zhang, Y., Jiang, H.* and Chen, P.R.* (2019). Sci. China Life Sci. 62, 1459-1471.
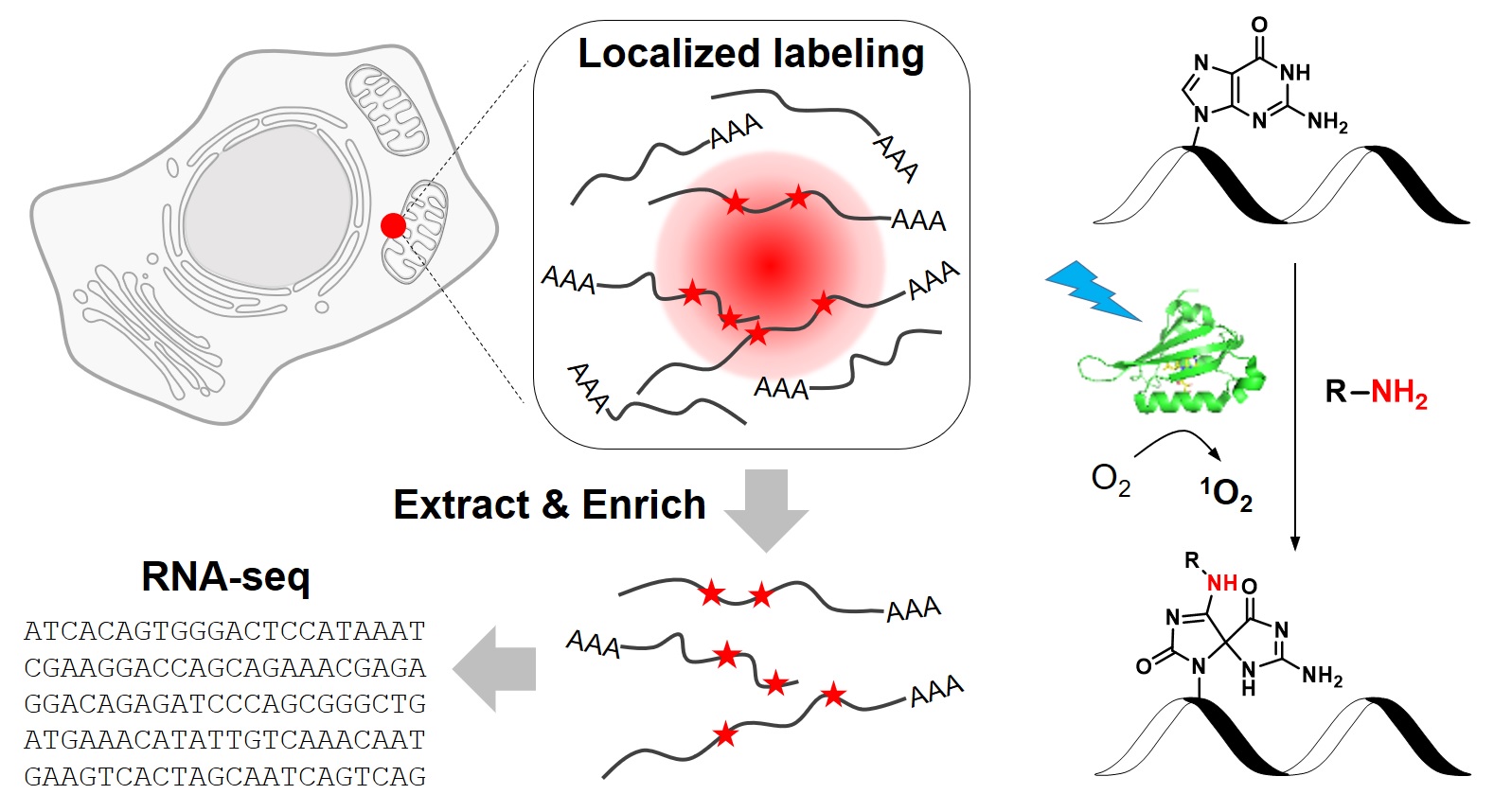
RNA molecules are highly compartmentalized in eukaryotic cells, with their localizations intimately linked to their functions. Despite the importance of RNA targeting, our current knowledge of the spatial organization of transcriptome has been limited by a lack of analytical tools. In this study, we develop a novel chemical biology approach to label RNAs in live cells with high spatial specificity. Our method, called CAP-seq, capitalizes on light-activated, proximity-dependent photo-oxidation of RNA nucleobases, which could be subsequently enriched via affinity purification and identified by high-throughput sequencing. Using this technique, we investigate the local transcriptomes that are proximal to various subcellular compartments, including the endoplasmic reticulum and mitochondria. We discover that mRNAs encoding for ribosomal proteins and oxidative phosphorylation pathway proteins are highly enriched at the outer mitochondrial membrane. Due to its specificity and ease of use, CAP-seq is a generally applicable technique to investigate the spatial transcriptome in many biological systems.
Wang, P.#, Tang, W.#, Li, Z.#, Zou, Z., Zhou, Y., Li, R., Xiong, T., Wang, J.* and Zou, P.* (2019). Nat. Chem. Biol. 15, 1110-1119.
Patients with liver diseases often suffer from chronic itch, yet the pruritogen(s) and receptor(s) remain largely elusive. Here, we identify bile acids as natural ligands for MRGPRX4. MRGPRX4 is expressed in human dorsal root ganglion (hDRG) neurons and co-expresses with itch receptor HRH1. Bile acids elicited Ca2+ responses in cultured hDRG neurons, and bile acids or a MRGPRX4 specific agonist induced itch in human subjects. However, a specific agonist for another bile acid receptor TGR5 failed to induce itch in human subjects and we find that human TGR5 is not expressed in hDRG neurons. Finally, we show positive correlation between cholestatic itch and plasma bile acids level in itchy patients and the elevated bile acids is sufficient to activate MRGPRX4. Taken together, our data strongly suggest that MRGPRX4 is a novel bile acid receptor that likely underlies cholestatic itch in human, providing a promising new drug target for anti-itch therapies.
Yu, H., Zhao, T., Liu, S., Wu, Q., Johnson, O., Wu, Z., Zhuang, Z., Shi, Y., Peng, L., He, R., Yang, Y., Sun, J., Wang, X., Xu, H., Zeng, Z., Zou, P., Lei, X., Luo, W.* and Li, Y.* (2019). Elife 8, e48431.
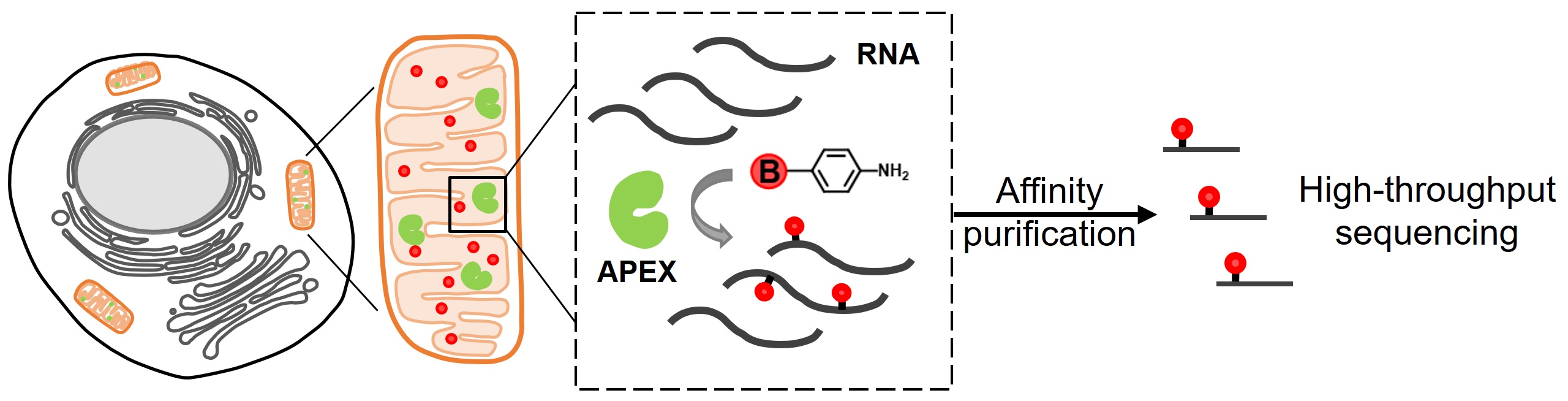
The subcellular organization of biomolecules such as proteins and nucleic acids is intimately linked to their biological functions. APEX2, an engineered ascorbate peroxidase that enables proximity-dependent labeling of proteins in living cells, has emerged as a powerful tool for deciphering the molecular architecture of various subcellular structures. However, only phenolic compounds have thus far been employed as APEX2 substrates, and the resulting phenoxyl radicals preferentially react with electron-rich amino acid residues. This narrow scope of substrates could potentially limit the application of APEX2. In this study, we screened a panel of aromatic compounds and identified biotin-conjugated arylamines as novel probes with significantly higher reactivity towards nucleic acids. As a demonstration of the spatial specificity and depth of coverage in mammalian cells, we applied APEX2 labeling with biotin-aniline (Btn-An) in the mitochondrial matrix, capturing all 13 mitochondrial messenger RNAs and none of the cytoplasmic RNAs. APEX2-mediated Btn-An labeling of RNA is thus a promising method for mapping the subcellular transcriptome, which could shed light on their functions in cell physiology.
Zhou, Y.#, Wang, G.#, Wang, P., Li, Z., Yue, T., Wang, J. and Zou, P.* (2019). Angew. Chem. Int. Ed. Engl. 58, 11763-11767.
In living organisms, protein functions are constantly evolving over generations throughout the history. Through iterative rounds of genetic mutations and natural selection of fit phenotypes, protein functions have been gradually optimized. This process could be mimicked and even greatly accelerated in the laboratory, when the selection pressure is directly applied to biomolecules of interest, which forms the basis of a technique called directed evolution. The Nobel Prize in chemistry 2018 was awarded jointly to Frances Arnold, George Smith and Gregory P. Winter for their pioneering contributions to the development and applications of directed evolution. Here we briefly review the history of this technique and its impact on renewable energy and pharmaceutical industry.
Zhou, Y.#, Zhu, L.# and Zou, P.* (2019). Univ. Chem. 34, 1-6.
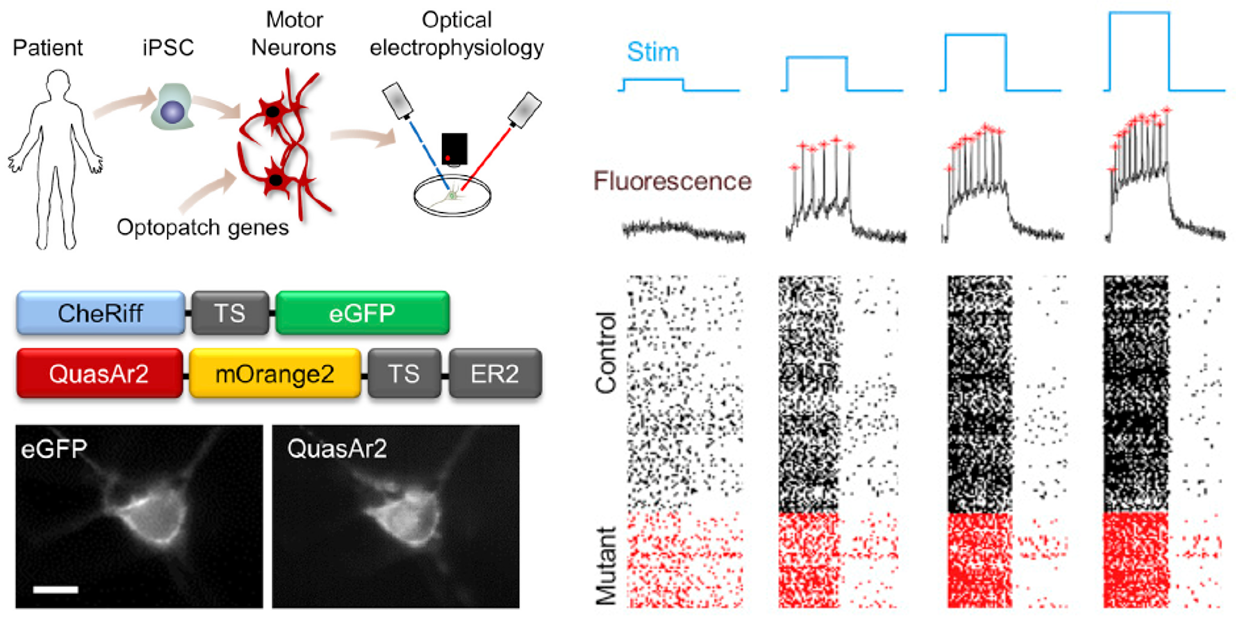
Human induced pluripotent stem cell (iPSC)-derived neurons are an attractive substrate for modeling disease, yet the heterogeneity of these cultures presents a challenge for functional characterization by manual patch-clamp electrophysiology. Here, we describe an optimized all-optical electrophysiology, "Optopatch," pipeline for high-throughput functional characterization of human iPSC-derived neuronal cultures. We demonstrate the method in a human iPSC-derived motor neuron (iPSC-MN) model of amyotrophic lateral sclerosis (ALS). In a comparison of iPSC-MNs with an ALS-causing mutation (SOD1 A4V) with their genome-corrected controls, the mutants showed elevated spike rates under weak or no stimulus and greater likelihood of entering depolarization block under strong optogenetic stimulus. We compared these results with numerical simulations of simple conductance-based neuronal models and with literature results in this and other iPSC-based models of ALS. Our data and simulations suggest that deficits in slowly activating potassium channels may underlie the changes in electrophysiology in the SOD1 A4V mutation.
Kiskinis, E.#, Kralj, J. M.#, Zou, P.#, Weinstein, E. N.#, Zhang, H., Tisoras, K., Wiskow, O., Ortega, J. A., Eggan, K.* and Cohen, A. E.* (2018). Stem Cell Reports 10, 1991-2004.
Optical biosensors have been invaluable tools in neuroscience research, as they provide the ability to directly visualize neural activity in real time, with high specificity, and with exceptional spatial and temporal resolution. Notably, a majority of these sensors are based on fluorescent protein scaffolds, which offer the ability to target specific cell types or even subcellular compartments. However, fluorescent proteins are intrinsically bulky tags, often insensitive to the environment, and always require excitation light illumination. To address these limitations, there has been a proliferation of alternative sensor scaffolds developed in recent years, including hybrid sensors that combine the advantages of synthetic fluorophores and genetically encoded protein tags, as well as bioluminescent probes. While still in their early stage of development as compared with fluorescent protein-based sensors, these novel probes have offered complementary solutions to interrogate various aspects of neuronal communication, including transmitter release, changes in membrane potential, and the production of second messengers. In this Review, we discuss these important new developments with a particular focus on design strategies.
Wang, A.#, Feng, J.#, Li, Y.* and Zou, P.* (2018). ACS. Chem. Neurosci. 9, 639-650.
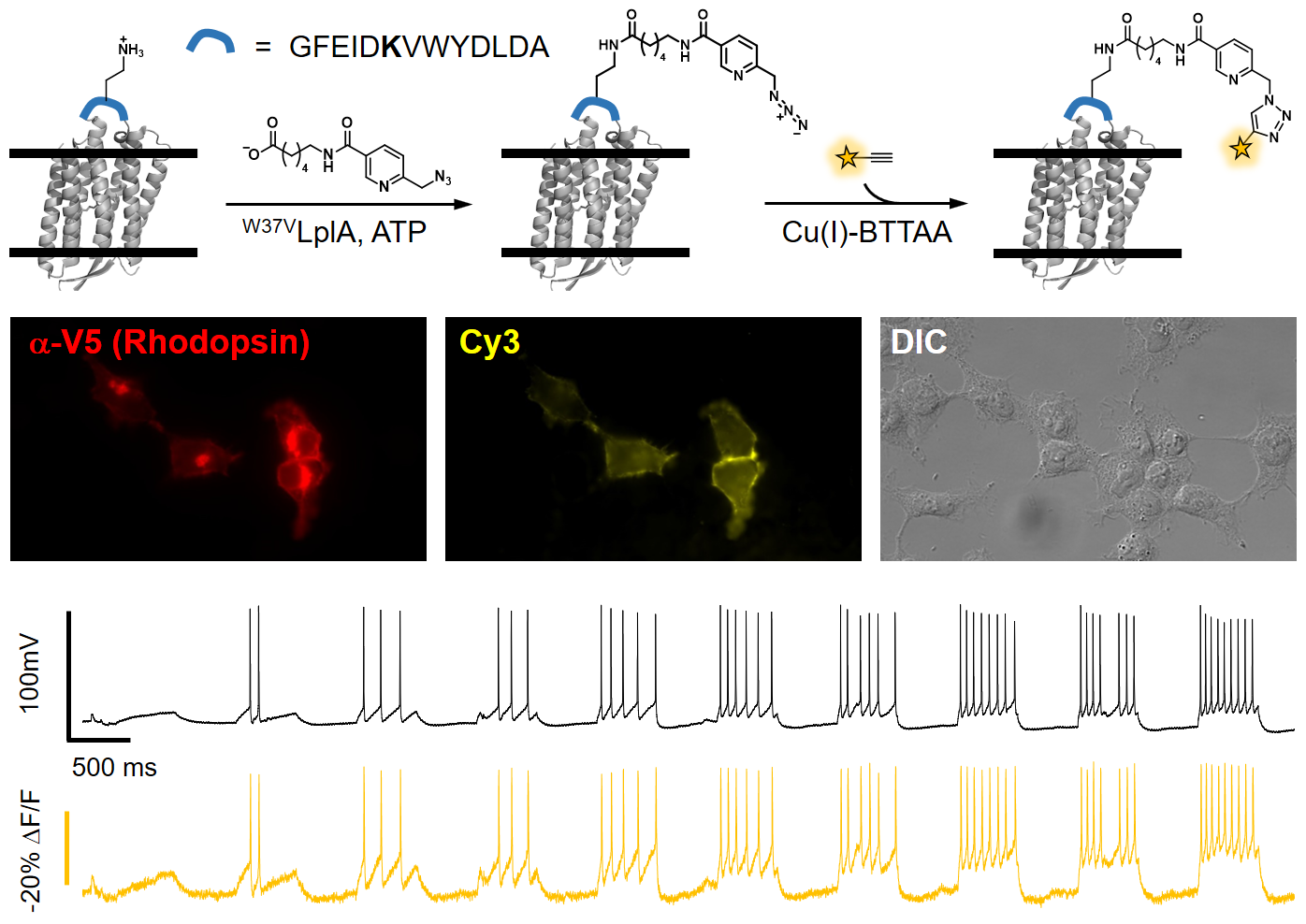
Membrane voltage is an important biophysical signal that underlies intercellular electrical communications. In this study, we present a novel fluorescent voltage indicator that enables the investigation of electrical signaling at high spatial resolution. Our method is built upon the site-specific modification of microbial rhodopsin proteins with organic fluorophores, resulting in a hybrid indicator scaffold that represents one of the most sensitive and fastest orange-colored voltage indicators developed to date. We applied this technique to optically map electrical connectivity in cultured cells, which revealed gap junction-mediated long-range coupling that spanned over hundreds of micrometers.
Xu, Y.#, Peng, L.#, Wang, S.#, Wang, A.#, Ma, R., Zhou, Y., Yang, J., Sun, D. E., Lin, W., Chen, X. and Zou, P.* (2018). Angew. Chem. Int. Ed. Engl. 57, 3949-3553.
We developed membrane voltage nanosensors that are based on inorganic semiconductor nanoparticles. We provide here a feasibility study for their utilization. We use a rationally designed peptide to functionalize the nanosensors, imparting them with the ability to self-insert into a lipid membrane with a desired orientation. Once inserted, these nanosensors could sense membrane potential via the quantum confined Stark effect, with a single-particle sensitivity. With further improvements, these nanosensors could potentially be used for simultaneous recording of action potentials from multiple neurons in a large field of view over a long duration and for recording electrical signals on the nanoscale, such as across one synapse.
Park, K., Kuo, Y., Shvadchak, V., Ingargiola, A., Dai, X., Hsiung, L., Kim, W., Zhou, H., Zou, P., Levine, A. J., Li, J. and Weiss, S.* (2018). Sci. Adv. 4, e1601453.
A holy grail in neuroscience is to understand how brain functions arise from neural network-level electrical activities. Voltage imaging allows for the direct visualization of electrical signaling at high spatial and temporal resolutions across a large neuronal population. Central to this technique is a palette of genetically-encoded fluorescent probes with fast and sensitive voltage responses. In this review, we chronicle the development and applications of genetically-encoded voltage indicators (GEVIs) over the past two decades, with a primary focus on the structural design that harness the power of fluctuating transmembrane electric fields. We hope this article will inform chemical biologists and protein engineers of the GEVI history and inspire novel design ideas.
Peng, L.#, Xu, Y.# and Zou, P.* (2017). Chin. Chem. Lett. 28, 1925-1928.

Membrane voltages are ubiquitous throughout cell biology. Voltage is most commonly associated with excitable cells such as neurons and cardiomyocytes, although many other cell types and organelles also support electrical signaling. Voltage imaging in vivo would offer unique capabilities in reporting the spatial pattern and temporal dynamics of electrical signaling at the cellular and circuit levels. Voltage is not directly visible, and so a longstanding challenge has been to develop genetically encoded fluorescent voltage indicator proteins. Recent advances have led to a profusion of new voltage indicators, based on different scaffolds and with different tradeoffs between voltage sensitivity, speed, brightness, and spectrum. In this review, we describe recent advances in design and applications of genetically-encoded voltage indicators (GEVIs). We also highlight the protein engineering strategies employed to improve the dynamic range and kinetics of GEVIs and opportunities for future advances.
Xu, Y., Zou, P.* and Cohen, A. E.* (2017). Curr. Opin. Chem. Biol. 39, 1-10.
Optical imaging of voltage indicators based on green fluorescent proteins (FPs) or archaerhodopsin has emerged as a powerful approach for detecting the activity of many individual neurons with high spatial and temporal resolution. Relative to green FP-based voltage indicators, a bright red-shifted FP-based voltage indicator has the intrinsic advantages of lower phototoxicity, lower autofluorescent background, and compatibility with blue-light-excitable channelrhodopsins. Here, we report a bright red fluorescent voltage indicator (fluorescent indicator for voltage imaging red; FlicR1) with properties that are comparable to the best available green indicators. To develop FlicR1, we used directed protein evolution and rational engineering to screen libraries of thousands of variants. FlicR1 faithfully reports single action potentials (~3% ΔF/F) and tracks electrically driven voltage oscillations at 100 Hz in dissociated Sprague Dawley rat hippocampal neurons in single trial recordings. Furthermore, FlicR1 can be easily imaged with wide-field fluorescence microscopy. We demonstrate that FlicR1 can be used in conjunction with a blue-shifted channelrhodopsin for all-optical electrophysiology, although blue light photoactivation of the FlicR1 chromophore presents a challenge for applications that require spatially overlapping yellow and blue excitation.
Abdelfattah, A. S., Farhi, S. L., Zhao, Y., Brinks, D., Zou, P., Ruangkittisakul, A., Platisa, J., Pieribone, V. A., Ballanyi, K., Cohen, A. E. and Campbell, R. E.* (2016). J Neurosci. 36, 2458-2472.
Before 2015
All-optical electrophysiology—spatially resolved simultaneous optical perturbation and measurement of membrane voltage—would open new vistas in neuroscience research. We evolved two archaerhodopsin-based voltage indicators, QuasAr1 and QuasAr2, which show improved brightness and voltage sensitivity, have microsecond response times and produce no photocurrent. We engineered a channelrhodopsin actuator, CheRiff, which shows high light sensitivity and rapid kinetics and is spectrally orthogonal to the QuasArs. A coexpression vector, Optopatch, enabled cross-talk–free genetically targeted all-optical electrophysiology. In cultured rat neurons, we combined Optopatch with patterned optical excitation to probe back-propagating action potentials (APs) in dendritic spines, synaptic transmission, subcellular microsecond-timescale details of AP propagation, and simultaneous firing of many neurons in a network. Optopatch measurements revealed homeostatic tuning of intrinsic excitability in human stem cell–derived neurons. In rat brain slices, Optopatch induced and reported APs and subthreshold events with high signal-to-noise ratios. The Optopatch platform enables high-throughput, spatially resolved electrophysiology without the use of conventional electrodes.
Hochbaum, D. R.#, Zhao, Y.#, Farhi, S. L., Klapoetke, N., Werley, C. A., Kapoor, V., Zou, P., Kralj, J. M., Maclaurin, D., Smedemark-Margulies, N., Saulnier, J. L., Boulting, G. L., Straub, C., Cho, Y. K., Melkonian, M., Wong, G. K. S., Harrison, D. J., Murthy, V. N., Sabatini, B. L., Boyden, E. S., Campbell, R. E. and Cohen, A. E.* (2014). Nat. Methods 11, 825-833.
Genetically encoded fluorescent reporters of membrane potential promise to reveal aspects of neural function not detectable by other means. We present a palette of multicoloured brightly fluorescent genetically encoded voltage indicators with sensitivities from 8–13% ΔF/F per 100 mV, and half-maximal response times from 4–7 ms. A fluorescent protein is fused to an archaerhodopsin-derived voltage sensor. Voltage-induced shifts in the absorption spectrum of the rhodopsin lead to voltage-dependent nonradiative quenching of the appended fluorescent protein. Through a library screen, we identify linkers and fluorescent protein combinations that report neuronal action potentials in cultured rat hippocampal neurons with a single-trial signal-to-noise ratio from 7 to 9 in a 1 kHz imaging bandwidth at modest illumination intensity. The freedom to choose a voltage indicator from an array of colours facilitates multicolour voltage imaging, as well as combination with other optical reporters and optogenetic actuators.
Zou, P.#, Zhao, Y. #, Douglass, A. D., Hochbaum, D. R., Brinks, D., Werley, C. A., Harrison, D. J., Campbell, R. E.* and Cohen, A. E.* (2014). Nat. Commun. 5, 4625.
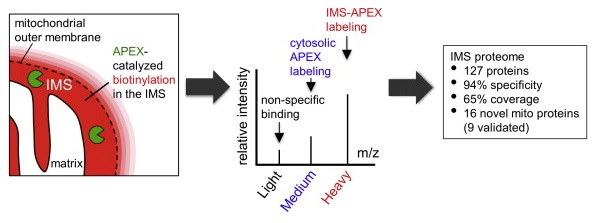
Obtaining complete protein inventories for subcellular regions is a challenge that often limits our understanding of cellular function, especially for regions that are impossible to purify and are therefore inaccessible to traditional proteomic analysis. We recently developed a method to map proteomes in living cells with an engineered peroxidase (APEX) that bypasses the need for organellar purification when applied to membrane-bound compartments; however, it was insufficiently specific when applied to unbounded regions that allow APEX-generated radicals to escape. Here, we combine APEX technology with a SILAC-based ratiometric tagging strategy to substantially reduce unwanted background and achieve nanometer spatial resolution. This is applied to map the proteome of the mitochondrial intermembrane space (IMS), which can freely exchange small molecules with the cytosol. Our IMS proteome of 127 proteins has >94% specificity and includes nine newly discovered mitochondrial proteins. This approach will enable scientists to map proteomes of cellular regions that were previously inaccessible.
Hung, V., Zou, P., Rhee, H., Udeshi, N. D., Cracan, V., Svinkina, T., Carr, S. A., Mootha, V. K. and Ting, A. Y.* (2014). Mol. Cell 55, 332-341.
The low-density lipoprotein receptor (LDLR) is a critical determinant of plasma cholesterol levels that internalizes lipoprotein cargo via clathrin-mediated endocytosis. Here, we show that the E3 ubiquitin ligase IDOL stimulates a previously unrecognized, clathrin-independent pathway for LDLR internalization. Real-time single-particle tracking and electron microscopy reveal that IDOL is recruited to the plasma membrane by LDLR, promotes LDLR internalization in the absence of clathrin or caveolae, and facilitates LDLR degradation by shuttling it into the multivesicular body (MVB) protein-sorting pathway. The IDOL-dependent degradation pathway is distinct from that mediated by PCSK9 as only IDOL employs ESCRT (endosomal-sorting complex required for transport) complexes to recognize and traffic LDLR to lysosomes. Small interfering RNA (siRNA)-mediated knockdown of ESCRT-0 (HGS) or ESCRT-I (TSG101) components prevents IDOL-mediated LDLR degradation. We further show that USP8 acts downstream of IDOL to deubiquitinate LDLR and that USP8 is required for LDLR entry into the MVB pathway. These results provide key mechanistic insights into an evolutionarily conserved pathway for the control of lipoprotein receptor expression and cellular lipid uptake.
Scotti, E., Calamai, M., Goulbourne, C. N., Zhang, L., Hong, C., Lin, R. R., Choi, J., Pilch, P. F., Fong, L. G., Zou, P., Ting, A. Y., Pavone, F. S., Young, S. G. and Tontonoz, P.* (2013). Mol. Cell. Biol. 33, 1503-1514.
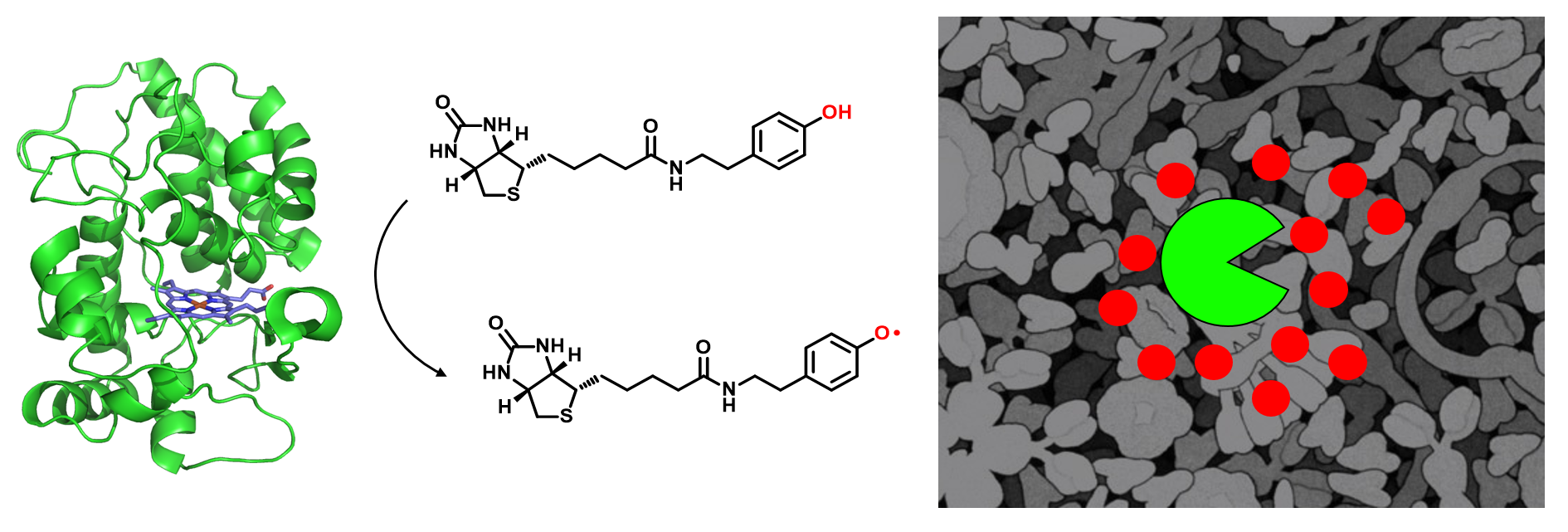
Microscopy and mass spectrometry (MS) are complementary techniques: The former provides spatiotemporal information in living cells, but only for a handful of recombinant proteins at a time, whereas the latter can detect thousands of endogenous proteins simultaneously, but only in lysed samples. Here, we introduce technology that combines these strengths by offering spatially and temporally resolved proteomic maps of endogenous proteins within living cells. Our method relies on a genetically targetable peroxidase enzyme that biotinylates nearby proteins, which are subsequently purified and identified by MS. We used this approach to identify 495 proteins within the human mitochondrial matrix, including 31 not previously linked to mitochondria. The labeling was exceptionally specific and distinguished between inner membrane proteins facing the matrix versus the intermembrane space (IMS). Several proteins previously thought to reside in the IMS or outer membrane, including protoporphyrinogen oxidase, were reassigned to the matrix by our proteomic data and confirmed by electron microscopy. The specificity of peroxidase-mediated proteomic mapping in live cells, combined with its ease of use, offers biologists a powerful tool for understanding the molecular composition of living cells.
Rhee, H. W.#, Zou, P.#, Udeshi, N. D., Martell, J. D., Mootha, V. K., Carr, S. A. and Ting, A. Y.* (2013). Science 339, 1328-1331.
A screen of Trp37 mutants of Escherichia coli lipoic acid ligase (LplA) revealed enzymes capable of ligating an aryl-aldehyde or aryl-hydrazine substrate to LplA's 13-residue acceptor peptide. Once site-specifically attached to recombinant proteins fused to this peptide, aryl-aldehydes could be chemoselectively derivatized with hydrazine-probe conjugates, and aryl-hydrazines could be derivatized in an analogous manner with aldehyde-probe conjugates. Such two-step labeling was demonstrated for AlexaFluor568 targeting to monovalent streptavidin in vitro, and to neurexin-1β on the surface of living mammalian cells. To further highlight this technique, we labeled the low-density lipoprotein receptor on the surface of live cells with fluorescent phycoerythrin protein to allow single-molecule imaging and tracking over time.
Cohen, J. D., Zou, P. and Ting, A. Y.* (2012). Chembiochem 13, 888-894.
The C-terminal domain (Mpro-C) of SARS-CoV main protease adopts two different fold topologies, a monomer and a 3D domain-swapped dimer. Here, we report that Mpro-C can reversibly interconvert between these two topological states under physiological conditions. Although the swapped α1-helix is fully buried inside the protein hydrophobic core, the interconversion of Mpro-C is carried out without the hydrophobic core being exposed to solvent. The 3D domain swapping of Mpro-C is activated by an order-to-disorder transition of its C-terminal α5-helix foldon. Unfolding of this foldon promotes self-association of Mpro-C monomers and functions to mediate the 3D domain swapping, without which Mpro-C can no longer form the domain-swapped dimer. Taken together, we propose that there exists a special dimeric intermediate enabling the protein core to unpack and the α1-helices to swap in a hydrophobic environment, which minimizes the energy cost of the 3D domain-swapping process.
Kang, X., Zhong, N., Zou, P., Zhang, S. N., Jin, C. W. and Xia, B.* (2012). Proc. Natl. Acad. Sci. U. S. A. 109, 14900-14905.
Methods to probe receptor oligomerization are useful to understand the molecular mechanisms of receptor signaling. Here we report a fluorescence imaging method to determine receptor oligomerization state in living cells during endocytic internalization. The wild-type receptor is co-expressed with an internalization-defective mutant, and the internalization kinetics of each are independently monitored. If the receptor internalizes as an oligomer, then the wild-type and mutant isoforms will mutually influence each others’ trafficking properties, causing co-internalization of the mutant or co-retention of the wild-type at the cell surface. Using this approach, we found that the low density lipoprotein (LDL) receptor internalizes as an oligomer into cells, both in the presence and absence of LDL ligand. The internalization kinetics of the wild-type receptor are not changed by LDL binding. We also found that the oligomerization domain of the LDL receptor is located in its cytoplasmic tail.
Zou, P. and Ting, A. Y.* (2011). ACS Chem. Biol. 6, 308-313.
Heparin-binding EGF-like growth factor (HB-EGF) is a ligand for EGF receptor (EGFR) and possesses the ability to signal in juxtacrine, autocrine and/or paracrine mode, with these alternatives being governed by the degree of proteolytic release of the ligand. Although the spatial range of diffusion of released HB-EGF is restricted by binding heparan-sulfate proteoglycans (HSPGs) in the extracellular matrix and/or cellular glycocalyx, ascertaining mechanisms governing non-released HB-EGF localization is also important for understanding its effects. We have employed a new method for independently tracking the localization of the extracellular EGF-like domain of HB-EGF and the cytoplasmic C-terminus. A striking observation was the absence of the HB-EGF transmembrane pro-form from the leading edge of COS-7 cells in a wound-closure assay; instead, this protein localized in regions of cell-cell contact. A battery of detailed experiments found that this localization derives from a trans interaction between extracellular HSPGs and the HB-EGF heparin-binding domain, and that disruption of this interaction leads to increased release of soluble ligand and a switch in cell phenotype from juxtacrine-induced growth inhibition to autocrine-induced proliferation. Our results indicate that extracellular HSPGs serve to sequester the transmembrane pro-form of HB-EGF at the point of cell-cell contact, and that this plays a role in governing the balance between juxtacrine versus autocrine and paracrine signaling.
Prince, R. N., Schreiter, E. R., Zou, P., Wiley, H. S., Ting, A. Y., Lee, R. T. and Lauffenburger, D. A.* (2010). J. Cell. Sci. 123, 2308-2318.
SARS coronavirus main protease (Mpro) plays an essential role in the extensive proteolytic processing of the viral polyproteins (pp1a and pp1ab), and it is an important target for anti-SARS drug development. We have reported that both the Mpro C-terminal domain alone (Mpro-C) and the N-finger deletion mutant of Mpro (Mpro-Δ7) exist as a stable dimer and a stable monomer (Zhong et al., J Virol 2008; 82:4227-4234). Here, we report structures of both Mpro-C monomer and dimer. The structure of the Mpro-C monomer is almost identical to that of the C-terminal domain in the crystal structure of Mpro. Interestingly, the Mpro-C dimer structure is characterized by 3D domain-swapping, in which the first helices of the two protomers are interchanged and each is enwrapped by four other helices from the other protomer. Each folding subunit of the Mpro-C domain-swapped dimer still has the same general fold as that of the Mpro-C monomer. This special dimerization elucidates the structural basis for the observation that there is no exchange between monomeric and dimeric forms of Mpro-C and Mpro-Δ7.
Zhong, N.#, Zhang, S. N.#, Xue, F.#, Kang, X., Zou, P., Chen, J. X., Liang, C., Rao, Z. H., Jin, C. W., Lou, Z. Y. and Xia, B. (2009). Protein Sci. 18, 839-844.
The main protease (Mpro) of severe acute respiratory syndrome coronavirus (SARS-CoV) plays an essential role in the extensive proteolytic processing of the viral polyproteins (pp1a and pp1ab), and it is an important target for anti-SARS drug development. It was found that SARS-CoV Mpro exists in solution as an equilibrium of both monomeric and dimeric forms, and the dimeric form is the enzymatically active form. However, the mechanism of SARS-CoV Mpro dimerization, especially the roles of its N-terminal seven residues (N-finger) and its unique C-terminal domain in the dimerization, remain unclear. Here we report that the SARS-CoV Mpro C-terminal domain alone (residues 187 to 306; Mpro-C) is produced in Escherichia coli in both monomeric and dimeric forms, and no exchange could be observed between them at room temperature. The Mpro-C dimer has a novel dimerization interface. Meanwhile, the N-finger deletion mutant of SARS-CoV Mpro also exists as both a stable monomer and a stable dimer, and the dimer is formed through the same C-terminal-domain interaction as that in the Mpro-C dimer. However, no C-terminal domain-mediated dimerization form can be detected for wild-type SARS-CoV Mpro. Our study results help to clarify previously published controversial claims about the role of the N-finger in SARS-CoV Mpro dimerization. Apparently, without the N-finger, SARS-CoV Mpro can no longer retain the active dimer structure; instead, it can form a new type of dimer which is inactive. Therefore, the N-finger of SARS-CoV Mpro is not only critical for its dimerization but also essential for the enzyme to form the enzymatically active dimer.
Zhong, N., Zhang, S. N., Zou, P., Chen, J. X., Kang, X., Li, Z., Liang, C., Jin, C. W. and Xia, B.* (2008). J. Virol. 82, 4227-4234.
Links
About Us
Peking University, Beijing, 100871, China
Tel: +86-10-62745395
Email: zoupeng_at_pku.edu.cn
ZouLab © All Rights Reserved.